
Testis of Male Tilapia Under Methomyl Stress: Transcriptome Changes and Signal Pathway Analysis
2020-08-14 10:16ShunlongMENGTaoLIUChaoSONGCongZHANGJiazhangCHEN
农业生物技术(英文版) 2020年2期
Shunlong MENG Tao LIU Chao SONG Cong ZHANG Jiazhang CHEN

Abstract This study was conducted to find the differentially expressed genes and understand the transcriptome change characteristics in the testis of tilapia (Oreochromis niloticus) stressed by methomyl. The tilapia were exposed to 200 μg/L methomyl for 30 d, and then the testis samples were collected and used for the high-throughput sequencing. The results showed that 369 significant differentially expressed genes were obtained, including 160 significantly down-regulated genes, and 209 significantly up-regulated genes. The enrichment of the differentially expressed genes in ECM-receptor interaction signaling pathway and MAPK signaling pathway was obvious. The adhesion of testis tissue cells was influenced by methomyl stress, which induced the expression of related gene in ECM-receptor interaction up-regulating, such as laminins, integrin α6 and integrin β1, in order to stabilize testis tissue structure. JNK, MAP3K and MKK4 in the JNK pathway were significantly up-regulated, which enhanced transcription factor AP-1 to promote the expression of P53, Bax and other pro-apoptotic proteins. The expression of growth-related factors in the ERK pathway of MAPK signal pathway was influenced by methomyl stress. The apoptosis-related genes and pregnancy-related genes have significant differential expression under methomyl stress, and thereby, the proliferation of reproduction cells in the testis tissue may be affected.
Key words Methomyl; Tilapia; Testis; Transcriptome; Signal pathway
Methomyl is a broad-spectrum carbamate-type pesticide, which has strong contact and stomach toxicity and has no intake transportation function. It is one of the most widely used pesticides. In 1997, the World Wildlife Fund listed methomyl as an estrogen-type endocrine disruptor[1]. Its biological toxicity to the environment is to destroy the predation relationship between animals and threaten the balance of ecosystems[2]. Due to the high solubility and large amount of use of methomyl in water, combined with the unreasonable discharge in industrial and agricultural production, some of the water and food residues are detected. At present, due to the high detection rate and endocrine disruption effect of methomyl in environmental waters, it has received extensive attention[3-5].
Transcriptome refers to all the transcripts and their corresponding quantity in cells, under specific development stages or physiological conditions. The transcription process uses a gene as the template to transcribe the precursor RNA through RNA polymerase, and then directly ligates the exons to form intact mature RNA by splicing between the spliceosomes[6]. Compared with traditional chip hybridization platforms, high-throughput RNA sequencing technology (RNA-Seq) can perform real-time detection of overall transcriptional changes without the need to design probes for known sequences, and has the characteristics of accurate digital signals, high detection throughput and more extensive detection scope[7]. At the single nucleotide level, RNA-Seq can detect transcriptional changes in any species and fully develop and analyze transcript expression in this physiological state[8]. These features make RNA-Seq a powerful tool for studying gene function and structure. RNA-Seq technology has been widely used to study gene expression analysis in specific tissues. In this study, the RNA of tilapia testis tissue was sequenced by the RNA-Seq high-throughput sequencing technology, so that the transcripts of tilapia testis under the stress of pesticide methomyl can be obtained, their expression levels can be reflected, and their location in gene pathways can be analyzed.
In this study, with male tilapia (Oreochromis niloticus) as a test species, the high-throughput RNA sequencing technology (RNA-Seq) was applied to study the transcriptome of tilapia testis under the action of pesticide methomyl, and the genes differentially expressed in the testis of tilapia under methomyl stress were obtained and further subjected to gene pathway analysis. This study will provide a basis for revealing the molecular mechanism of methomyls reproductive toxicity to male tilapia.
Materials and Methods
Materials
Male tilapia as a test species were taken from the Yixing breeding base of the Freshwater Fisheries Research Center of Chinese Academy of Fishery Science. The fish had an average body weight of (112.24±9.48) g and an average body length of (17.06±0.91) cm. Thirty male tilapia were randomly placed in an aquarium with a volume of 300 L (containing 200 L of water) and domesticated for 4 weeks. They were fed once daily at 8:00 during the domestication period, and the feeding amount was calculated according to 2% of the body weight of the tilapia fish. The ratio of dark to light was 12 h∶12 h. An air compressor was used for oxygenation.
Experimental water
The experiment water was dechlorinated tap water that had been aerated for 1 week. It had a temperature of (25±0.5) ℃ and a pH value of (7.0±0.5), and contained 6.3-7.0 mg/L of dissolved oxygen. The experiment water met the fishery water quality standard (GB 11607-89).
Experimental design
According to our previous acute test results (the 96-h LC50 of methomyl was 940 μg/L), the residual amount of methomyl in natural water (0-97 μg/L)[9-11] and the maximum limit of methomyl (200 μg/L) specified by the US water quality standards[12], five concentration gradients were set in this experiment: 0, 0.2, 2, 20 and 200 μg/L. Each concentration gradient was set up in 3 parallel groups. 200 L of each of the above concentrations of contamination solution was placed in a 300 L aquarium, and each parallel group was randomly added with 20 healthy tilapia. The experiment was carried out by semi-static water contact contamination method. The water in each water tank of each concentration was replaced by 50% every day, and the drug was added to the original concentration. The experiment lasted for 48 d, in which the first 30 d was the methomyl exposure phase, and the last 18 d was the clean water recovery phase (no methomyl). In order to obtain the actual content of methomyl in each concentration group, the actual concentrations were determined with reference to the methods provided by Zhang et al.[13] and Zhang et al.[14]. The methomyl contents of the 0, 0.2, 2, 20 and 200 μg/L groups were 0, 0.23, 2.12, 21.50 and 182.0 μg/L, respectively, at the beginning of exposure and were 0, 0.21, 1.92, 18.52, and 179.0 μg/L, respectively, 24 h later. The concentration of methomyl in each concentration group 24 h later was not much different from that at the beginning of exposure. The water in each tank of each concentration was replaced by 50% every day, and the semi-concentration methomyl was added to ensure the stability of methomyl.
Sample collection
Samples were collected from the 200 μg/L treatment group and the control group on the 30th d of exposure. Feeding was stopped 24 h before sampling. During the experiment, tilapia were active and healthy. Tilapia individuals were anesthetized with 250 mg/L MS-222 at the time of sampling, and the body length and body weight were measured. The testis samples were quickly collected after killing the fish. The testis samples were placed in a 1.5 ml RNase-free centrifuge tube, rapidly placed into liquid nitrogen, and frozen and stored in an ultra-low temperature freezer at -80 ℃.
RNA-Seq Data
The testis tissues placed in a -80 ℃ ultra-low temperature freezer were taken out and sent under ice-cooling conditions to Nanjing Decode Genomics Biotechnology Co., Ltd. for sequencing. The transcriptome sequencing results of the tilapia testis tissues were obtained by the high-throughput sequencing platform Seq2000, and gene differential expression analysis was performed.
Original data filtering
Sample contamination, joint contamination and sequencing errors all would affect the quality of sequencing results. In order to ensure the accuracy of the analysis results, quality control was performed on the Raw data, and the data was filtered. Then, data quality control was performed with FastQC (http://www. bioinformatics. babraham.ac.uk/projects/fastqc/), and the Illumina sequencing Reads quality assessment and analysis software Solexa QA were used to remove the joint-containing sequences; the sequences containing N at ratios greater than 10% were removed; the low-quality sequences were removed; and the data retained after filtering was clean reads for subsequent information analysis.
Annotation of clean reads
For clean reads obtained after filtration, Tophat[15] software was used to align the clean reads with the tilapia reference genome (Oreochromis_niloticus.Orenil1) (ftp://ftp.ensembl.org/pub/release- 84/fasta/ oreochromis_niloticus/dna/), under following alignment parameters: -segment-length 25-segmentmism-atches 2, maximum allowable number of mispairing 12, with other parameters defaulted.
Gene expression profiling
For the results of the Tophat alignment, HTSeq[16] software was used to statistically count the number of sequences aligned to each gene, with parameters defaulted. The edge R[17] (http://www.bioconductor.org/packages/release/bioc/html/edgeR.html) software package was then used to perform inter-group comparison to screen for differentially expressed genes. Edge R calculated the Fold Change and p-value of the genes between different groups. The p-value reflected the significance of the differential expression, and was further corrected to obtain FDR. The genes with FDR≤0.05 are significant differentially expressed genes. WEGO was used to classify differential genes by GO (gene ontology). They were classified into three categories: cell component, molecular function, and biological process. The differentially expressed genes were localized in cell signaling pathways by KEGG. Pathway enrichment analysis was performed using KOBAS with FDR≤0.05 as the standard of a significant enrichment signal pathway to determine significant enrichment signaling pathways involving the significant differentially expressed genes, to thereby study the roles and regulatory mechanisms of these genes in the organism.
Results and Analysis
Annotation of sequencing data
In this study, a total of 45 million filtered sequences were obtained from the two libraries, and these sequences were compared with the tilapia reference genome. The results are shown in Table 1. In each library, about 0.15 sequence was uniquely aligned to the genome, accounting for 65% to 66% of all original sequences, accounting for 98% of the total number of sequences.
Analysis of gene expression profiles
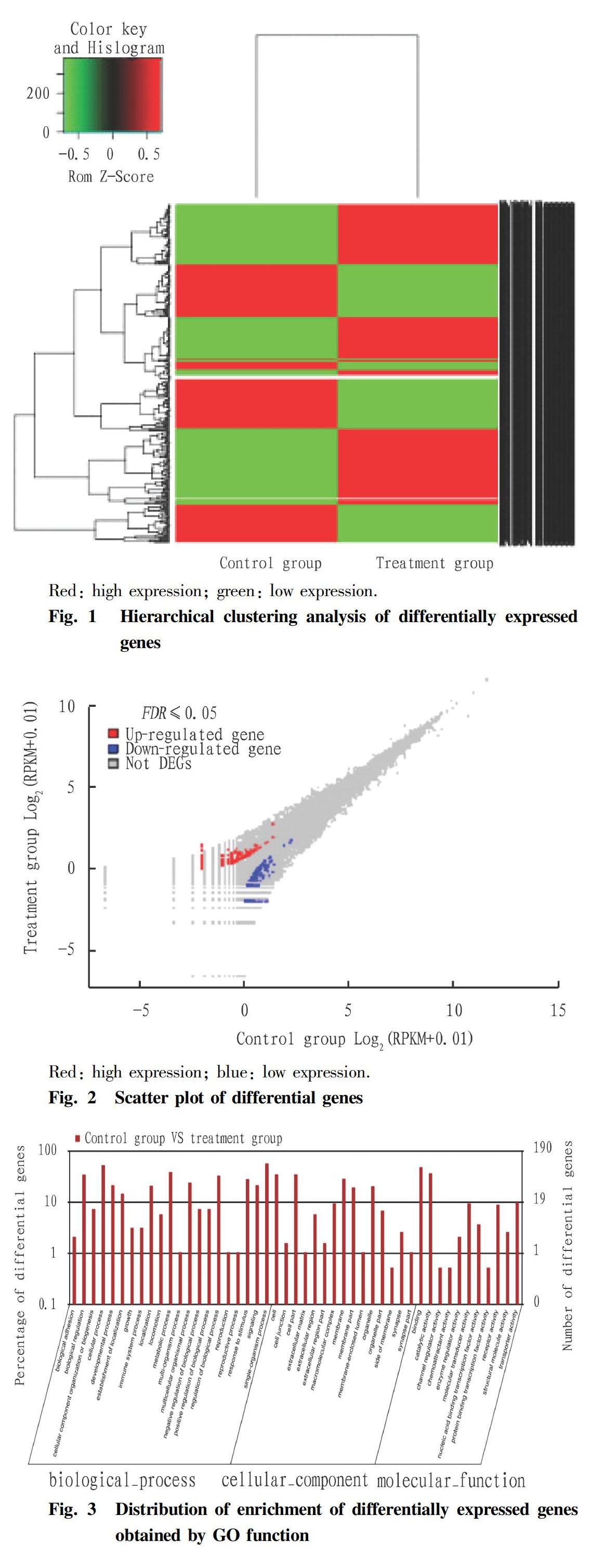
A total of 19 158 genes were expressed in two transcriptome libraries, of which 17 884 were expressed in the control group and 18 533 were expressed in the methomyl-treated group. There were 369 significantly expressed genes, of which 160 were down-regulated and 209 were up-regulated. Hierarchical clustering analysis (Fig. 1) and distribution scatter analysis (Fig. 2) were performed on all differentially expressed genes in the two libraries. The hierarchical clustering of the differential genes revealed that similar genes were expressed under different experimental conditions. The distribution scatter analysis was used to observe the expression levels and distribution of differential genes in different groups. The results showed that the gene expression profile of the testis tissue of the tilapia in the control group was significantly different from that of the testis tissue of the tilapia in the treatment group, which indicated that methomyl stress had an effect on the transcriptome of the tilapia testis.
The genes differentially expressed in the two libraries were classified into three categories: biological process, cellular component and molecular function (Fig. 3). In the category of biological process, most of the differential genes were concentrated in metabolic processes, cellular processes and single organism processes; in the category of cellular component, most of the differential genes were related to cell composition (cell, cell part) and membrane; and in the category of molecular function, genes related to binding and catalytic activity were the most.
Analysis on significance of gene expression differences
The Edge R software was used to analyze the differences in RNA expression levels between the control group and the treatment group. Finally, 369 genes differentially expressed in the tilapia testis under the methomyl stress were detected, including 160 significantly down-regulated genes and 209 significantly up-regulated genes. In this study, the differentially expressed genes were classified by GO function (Fig. 3). In the comparison between the control group and the methomyl-treated group, it was found that the genes whose expression was significantly up-regulated in the latter were mostly concentrated in the cell composition, biological process, catalytic activity, metabolic process and stress response categories. Genes whose expression was significantly down-regulated were also enriched in the metabolic process, cell composition and biological process categories.
Analysis on the enrichment signaling pathways of differentially expressed genes
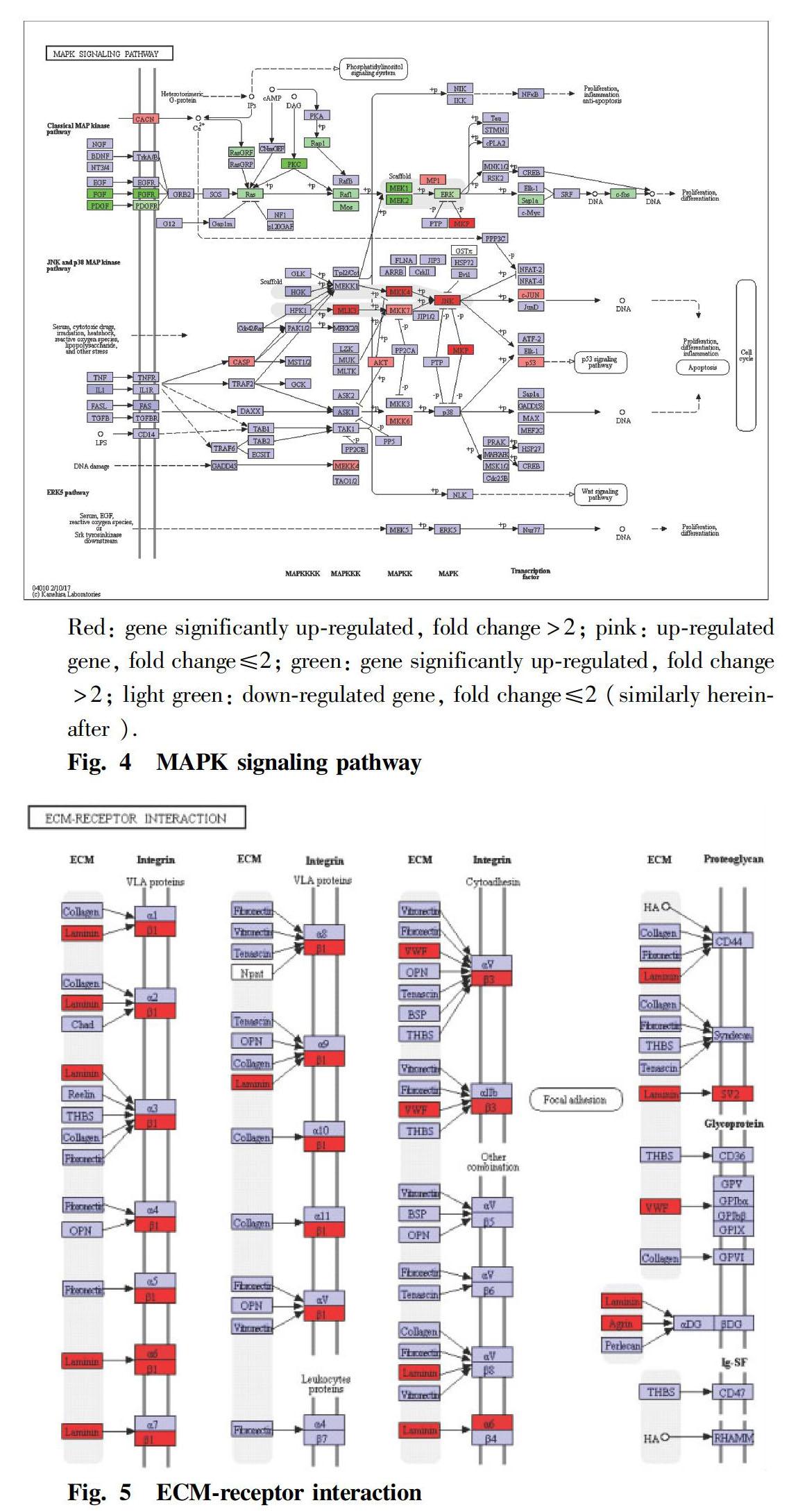
The genes differentially expressed in the testis of tilapia under methomyl stress were obtained, and they were mapped to the cellular signaling pathways by differentially expressed gene expression levels and G functional categories. The results showed that the significant differentially expressed genes in the tilapia testis tissues were significantly enriched in the MAPK signaling pathway and the between ECM-receptor interaction signaling pathway. In the Raf-Mek-Erk pathway of the MAP kinase pathway, Fgf1, Pdgfa, Fgfr and Pkc were significantly down-regulated, and the expression levels of related genes such as Ras, Raf1 and Mek were also down-regulated (Fig. 4). The expression of MAP3K, MKK4, JNK and P53 was up-regulated in the JNK pathway of the MAP kinase pathway. In the ECM-receptor interaction, the expression of Lam, Itgb1, Itgb3, Itga6, Vwf and the like was significantly up-regulated (Fig. 5).
Shunlong MENG et al. Testis of Male Tilapia Under Methomyl Stress: Transcriptome Changes and Signal Pathway Analysis
Discussion
The extracellular matrix (ECM) is a complex mixture of structural and functional macromolecules that plays an important role in cell and tissue structure and function maintenance and tissue and organ morphogenesis. ECM conducts signals through the transmembrane surface receptor integrins. Integrins are transmembrane glycoproteins, as well as a class of cell membrane surface receptors that recognize and bind to the corresponding ligands in the extracellular matrix, providing an attachment point for cell adhesion[18]. In the interaction between ECM and receptor integrins, the expression of laminins in ECM and integrins ɑ6, β1, and β3 were significantly up-regulated. Integrins β1 and ɑ6 are important adhesion molecules in the spermatogenic epithelium, which constitute heterodimers on the surface of the supporting cells[19], interact with the laminins on the surface of sperm cells, and participate in the top surface of the spermatogenic epithelium, while the spermatogenic cells are connected to the spermatogenic epithelium side via adhesion molecules[20]. In this study, high concentration of methomyl had an effect on the structure of tilapia testis by inducing integrin β1 to be significantly up-regulated; and laminins, integrins ɑ6 and β1 were up-regulated, which was probably because that methomyl damaged the cell adhesion of tilapia testis tissue, and the organism itself exhibited a feedback mechanism for stabilizing the testis tissue structure.
MAPK is a class of serine/threonine protein kinases that are widely found in mammals. MAPK can be activated by extracellular signals such as cellular stress, cytokines and cell adhesion. MAPK plays an important role in cell proliferation, differentiation, transformation and apoptosis, such as phospho-nuclear factors, enzymes and cytoskeletal proteins. The MAPK signaling pathway has four main pathways: protein kinase (ERK), c-Jun N-terminal kinase (JNK), P38MAPK, and ERK5. External stress such as oxidative damage and electrical radiation all may activate the JNK signaling pathway, which induces activation of MAP3Ks, activates MAP2K isomers MKK4 and MKK7, and finally phosphorylates JNK[21]. The JNK signaling pathway plays an important role in stress response, apoptosis and the occurrence of various pathological conditions, and many studies have shown that the JNK signaling pathway is closely related to apoptosis. Wang et al.[22] found that neuroblastoma was induced by methamphetamine to apoptosis, accompanied by a significant increase in the expression level of phosphorylated JNK, and the apoptosis was significantly reduced after inhibiting NK activity. Eilers et al.[23] found that the inhibition of JNK activity in sympathetic cells can reduce cell apoptosis. JNK phosphorylation promotes the expression of proapoptotic proteins such as P53 and Bax by enhancing the transcription factor AP-1[24]. The results of this study showed that MAP3K, MKK4 and JNK expression were significantly up-regulated in the JNK signaling pathway. The up-regulated expression of JNK enhanced the transcription factor AP-1, thereby promoting the expression of proapoptotic proteins such as P53 and Bax. Therefore, it could be seen that the stress of the environmental estrogen methomyl had an effect on the expression of JNK pathway-related genes in the MAPK signaling pathway of tilapia testis tissue, and their expression was significantly up-regulated. The reason might be the stress caused by methomyl toxicity stress. Meanwhile, studies have shown that methomyl can cause oxidative stress in rat testis tissue, tilapia liver, and blood[25-26]. It suggests that the up-regulation of JNK and P53 may also be related to oxidative stress.
The ERK pathway plays an important role in cell proliferation in the MAPK signaling pathway. In the ERK pathway, Fgf1 and Pdgfa act as upstream factors to initiate the entire Raf-Mek-Erk cascade reaction. In this study, the expression of Fgf1 and Pdgfa was significantly down-regulated. Both Fgf1 and Pdgfa belong to growth factors, and they bind to the receptors Pdgfr and Fgfr to activate tyrosine kinase. Meanwhile, the results showed that the expression levels of Pdgfr and Fgfr were also down-regulated. Some researchers have reported that Pdgfr can transfer the meiocytes in the center of the convoluted somniferous tubules of the reproductive tissue to the basement membrane of the spermatogenic epithelium and stimulate them to differentiate into spermatogonial stem cells[27]. Therefore, from the Pdgfr expression level, the number of spermatogonial cells in the testis tissue might be affected. The activated tyrosine kinase is capable of transmitting a signal to the Ras protein in the midstream of the cascade reaction. The Ras protein has the activity of GTPase. It binds to GTP to form Ras-GTP, which interacts with Raf1 to activate Raf-1, transmits cell proliferation signals from the cell membrane to the cytoplasm and nucleus, phosphorylates and activates a series of downstream kinases, and finally phosphorylates ERKs. The activated ERKs rapidly enter nuclear phosphorylation and activate transcriptional molecules involved in proliferative responses such as ELK-1 and C-fos, thereby promoting cell proliferation[28]. The results of this study showed that the expression of Fgf, Ras, Raf1, MEK1/2 and C-fos in the upstream, midstream and downstream parts of the Raf-Mek-Erk pathway were all significantly down-regulated. The ERK pathway in the MAPK signaling pathway is mainly regulated by growth factors. It could be seen that the expression of growth factors in the tilapia testis was affected by the stress of the environmental estrogen methomyl, which further affected the Raf-Mek-Erk cascade reaction in the MAPK signaling pathway and might have an effect on the proliferation of the germ cells of tilapia testis, resulting in a decrease in the number of spermatogenic cells and sperm.
Conclusions
Through the high-throughput sequencing analysis of the tilapia testis under the methomyl stress, 369 genes differentially expressed in the tilapia testis were detected, including 160 significantly down-regulated genes and 209 significantly up-regulated genes.
The gene expression profile of the tilapia testis tissue under the methomyl stress was significantly different from that of the control group, indicating that the methomyl stress had a significant effect on the transcriptome of the testis of tilapia.
The apoptosis-related genes and pregnancy-related genes have significant differential expression under methomyl stress, and thereby, the proliferation of reproduction cells in the testis tissue may be affected.
References
[1] GUO XB. Environmental health[M]. Beijing: Peking University Medical Press, 2006. (in Chinese)
[2] SUN L, LEE HK. Stability studies of propoxurher-bisice in environmental water samples by liquid chromatography[J]. Journal of Chromatography, 2003, 10(14): 153-163.
[3] MENG SL, QIU LP, HU GD, et al. Effects of methomyl on steroidogenic gene transcription of the hypothalamic- pituitary-gonad-liver axis in male tilapia[J].Chemosphere, 2016(165): 152-162.
[4] MENG SL, QIU LP, HU GD, et al. Responses and recovery pattern of sex steroid hormones in testis of Nile tilapia (Oreochromis niloticus) exposed to sublethal concentration of methomyl[J]. Ecotoxicology, 2016, 25(10): 1-7.
[5] MENG SL, QIU LP, HU GD, et al. Effect of methomyl on sex steroid hormone and vitellogenin levels in serum of male tilapia (Oreochromis niloticus) and recovery pattern[J]. Environmental Toxicology, 2017(32): 1869-1877.
[6] TIM R MERCER, IRFAN A QURESHI, SOLEN GOKHAN, et al. Long noncoding RNAs in neuronal-glial fate specification and oligodendrocyte lineage maturation[J]. BMC Neuroscience, 2010, 11(1): 14.
[7] MARCEL M, MICHAEL E, WILIAM EA, et al. Genome sequencing in open microfabricated high density picoliter reactors[J]. Nature, 2005, 437(7057): 376-380.
[8] ZHANG CL, WANG GZ, QIN ZJ, et al. Transcriptome and RNA-Seq technology[J]. Biotechnology Bulletin, 2012(12): 51-56. (in Chinese)
[9] VAN-SCOY AR, YUE M, DENG X, et al. Environmental fate and toxicology of methomyl[J].Reviews of environmental contamination and toxicology, 2013(222): 93-109.
[10] EL-SAEID MH, AL-TURKI AM, AL-WABLE MI, et al. Evaluation of pesticide residues in Saudi Arabia ground water[J].Research Journal of Environmental Sciences, 2011, 5(2): 171-178.
[11] WANG J, LIU ZZ, PAN HF, et al. Analytical method, pollution pattern and health risk of carbamate for the city raw water of Zhejiang[J]. Environmental Chemistry, 2010, 29(4): 623-628. (in Chinese)
[12] U.S.EPA. 2012 Edition of the drinking water standards and health advisories[M]. EPA 822- S- 12- 001. Office of Water U.S. Environmental ProtectionAgency, Washington, DC, 2012.
[13] ZHANG XJ, CHENG YH, LI SR. Determination of carbamate pesticides in drinking water using solid phase extract-HPLC[J]. Chinese Journal of Health Laboratory Technology, 2014, 24(9): 1246-1247. (in Chinese)
[14] ZHANG B, FU XQ, XU NB, et al. Determination of Carbamates and their metobolites in water using liquid chromatography-mass spectrometry[J]. Environmental Protection of Chemical Industry, 2010, 30(4): 364-367. (in Chinese)
[15] TRAPNELL C, PACHTER L, SALZBERG SL. Top Hat: discovering splice junctions with RNA-Seq[J]. Bioinformatics, 2009, 25(9): 1105-1111.
[16] ANDERS S, PYL PT, HUBER W. HTSeq-A Python framework to work with high- throughput sequencing data[J].Bioinformatics, 2015, 31(2): 166.
[17] ROBINSON MD, MCCARTHY DJ, SMYTH GK. edge R: a Bioconductor package for differential expression analysis of digital gene expression data[J]. Bioinformatics, 2010, 26(1): 139-140.
[18] GOODMAN SL, PICARD M. Integrins as therapeutic targets[J].Trends Pharmacol Sci, 2012, 33(7): 405-412.
[19] SALANOVA M, STEFANINI M, DE CURTIS I, et al. Integrin receptor alpha 6bata 1is localized at specific sites of cell-to-cell contact in rat seminiferous epitheium[J].Biol Reprod,1995,52(1):79-87.
[20] LEE NP, CHENG CY. Ectoplasmic specialization, a testis-specific cell-cell actin-based adherens junction type: is this a potential target for male contraceptive development[J].Hum Reprod Update,2004,10(4): 349-369.
[21] WESTON CR, DAVIS RJ. The JNK signal transduction pathway[J]. Curr Opin Genet Dev, 2002, 12(1): 14-21.
[22] WANG SF, YEN JC, YIN PH, et al. Involvement of oxidative stress-activated JNK signaling in the methamphetamine-induced cell death of human SH-SY5Y cells[J]. Toxicology, 2008, 246(2/3): 234-241.
[23] EILERS A, WHITFIELD J, SHAH B, et a1. Direct inhibition of cJun N-terminal kinase in sympathetic neurones prevents c-jnil promoter activation and NGF withdrawal-induced death[J]. J Neurochem.200l,76(5): 1439-1454.
[24] FU YY, LI YL. JNK signaling pathway and apoptosis[J]. Journal of Chongqing Medical University, 2014, 39(3): 281-283. (in Chinese)
[25] HU MM, LIU XF, GUAN X, et al. Joint action of phoxim and methomyl on female rats reproductive toxicity[J]. Carcinogenesis, Teratogenesis & Mutagenesis, 2008, 20(6): 470-474. (in Chinese)
[26] MENG SL. Effects of environmental estrogen methomyl on the hypothalamic-pituitary-gonadal axis and antioxidant defense system of male tilapia[D]. Nanjing: Nanjing Agriculture University, 2014. (in Chinese)
[27] BASCIANI S, MARIANI S, SPERA G, et al. Role of platelet-derived growth factors in the testis[J]. Endocr Rev, 2010(31): 916-939.
[28] BIOLLY B, VERCOUTER- EDOUART AS, HONDERMARK H, et al. FGF singnals for cell proliferation and migration through different pathways[J]. Cy-tokine Growth Factor Rev, 2000, 1(4): 295-302.

- 农业生物技术(英文版)的其它文章
- Enhanced Production of Natural Carotenoids from Genetically Engineered Rhodobacter sphaeroides Overexpressing CrtA
- Functional Analysis of Dunaliella salina Calmodulin Kinase Gene
- First Report of Phomopsis Leaf Spot on Patchouli [Pogostemon cablin (Blanco) Benth.] Caused by Diaporthe arecae in China
- Effects of Plant Growth Regulators on Tillering Ability of Ophiopogon japonicus cv
- Effect of Irrigation and Fertilization on Population Structure and Yield of Wheat
- Production and Cultivation Technology of Selenium-enriched Pueraria thomsonii Benth