
6957例新生儿听力筛查联合耳聋基因检测结果分析
2020-08-08 03:31:06阳彦刘艳秋罗海艳邹欢欢马鹏鹏
实用医学杂志 2020年14期
阳彦 刘艳秋 罗海艳 邹欢欢 马鹏鹏
江西省妇幼保健院产前诊断中心(南昌330006)
先天性耳聋是人类最常见的出生缺陷之一,是全球重大公共卫生问题,2014年WHO 估计,全球因聋致残人数高达3.6 亿,约占世界总人口5.14%[1]。新生儿听力筛查为全国新生儿重点筛查三大疾病筛查之一,2010年卫生部颁布了《新生儿听力筛查技术规范》,旨在尽早发现有听力障碍的个体,并及时给予适当干预,避免由聋致哑。随着新生儿听力筛查工作的深入进行,国内外研究均报道单纯听力筛查可能会漏诊迟发性耳聋患儿及药物性耳聋基因携带者。2007年,新生儿听力联合基因筛查的理念被首次提出[4]。近年来,新生儿听力与基因联合筛查越来越被医学界所关注,为耳聋三级预防的有效手段。
本研究联合新生儿听力筛查与耳聋基因芯片技术对6 957 例新生儿进行常见耳聋基因的检测,在前人研究[5-7]基础上,增加了对杂合携带者进行相应基因Sanger 测序,避免漏诊非热点突变的耳聋患儿,以达到降低耳聋发生率,同时初步明确江西地区非综合征型耳聋基因突变热点与突变谱,达到完善我国非综合征型耳聋分子流行病学数据的目的。
1 对象与方法
1.1 研究对象2011年11月至2019年6月江西省妇幼保健院产科出生的6 957 例正常活产新生儿,出生时Apgar 评分均为10 分,所有新生儿均为母婴同室,均无产科并发症,不包括伴听力损失高危因素的新生儿。父母均为江西籍贯。联合筛查前均进行充分知情告知并签署知情同意书。该研究符合2013 修订的《赫尔辛基宣言》(www.wma.net/en/30publications/10policies/b3/index.html)的要求,本研究通过江西省妇幼保健院伦理委员会的审批。
1.2 方法
1.2.1 听力筛查方法新生儿于出生3 d 后采用耳声发射法(otoacousticemission,OAE)进行初次听力筛查,初筛不通过者在出生后29~42 d 进行复筛,复筛采用OAE 与自动判别听性脑干诱发电位法(auto auditorybrainstemresponse,AABR)结合检测。复筛未通过者,于3 个月龄进行听性脑干反应(auditory brainstem response,ABR)进行诊断。
1.2.2 耳聋基因检测方法采取新生儿EDTA抗凝血1 mL,采用天隆NP968DNA 自动提取系统标准操作规程提取基因组DNA,采用NanoDrop2000 测定DNA 浓度与纯度,应用晶芯®9 项遗传性耳聋基因检测试剂盒(微阵列芯片法)对4 个耳聋基因的9 个位点进行检测,包括GJB2(c.235delC,c.299_300delAT,c.176_191del16 ,c.35delG)、SLC26A4(IVS7-2A>G,c.2168A>G)、线粒体12SrRNA(m.1555A>G,m.1494C>T)和GJB3(c.538C>T)。具体实验操作参见试剂盒说明书。
1.2.3 杂合突变携带者基因测序对耳聋基因芯片检测结果为杂合突变携带者进一步行相应基因测序,按相应扩增条件完成PCR 测序后送上海生工测序,部分引物序列见表1,序列分析软件为FinCH TV。
1.3 统计学方法建立EXCEL 数据库,采用SPSS 19.0进行χ2检验,P<0.05为差异有统计学意义。
2 结果
2.1 新生儿耳聋基因芯片检测结果共完成6 957例新生儿耳聋基因检测,检出耳聋基因突变新生儿282 例,其中GJB2 基因杂合突变142 例,GJB2 c.235delC 纯合突变1 例,双杂合突变3 例,分别为c.235delC/c.299_300 del AT双杂合、GJB2 c.235delC/c.109 A>G 双杂合、GJB2 c.235delC/m.1555A>G 均质突变以及1 例3 个突变的复合杂合突变GJB2c.235delC/SLC26A4IVS7-2A>G/SLC26A4 c.109G>T 。GJB2 c.35delG 杂合突变1 例(0.01%),GJB2 c.176_191del16 杂合突变7 例(0.1%),GJB2 c.299_300delAT 杂合突变28 例(0.4%)。发现GJB3 c.538C>T杂合突变7 例(0.1%),SLC26A4 杂合突变68 例,其中SLC26A4IVS7-2A>G 杂合突变52 例(0.74%),c.2168A>G杂合突变16例(0.22%),SLC26A4 IVS7-2A>G 复合c.1173C>A 1例(0.01%)。另发现线粒体基因突变26 例(0.37%),包括线粒体12SrRNA m.1555A>G 均质突变20 例(0.28%),1 例为GJB2 c.235delC/m.1555A>G 均质突变双杂合突变。m.1555A>G 异质突变3 例(0.04%),线粒体12S rRNA1494 C>T 均质突变1 例(0.01%),异质突变1 例(0.01%)。基因芯片检测原始数据见图1,基因突变类型及等位基因携带频率见表2,所有突变携带者均进行相应基因测序。
2.2 新生儿耳聋基因芯片检测杂合携带者测序结果282 例携带者中,除25 例单纯为线粒体突变携带者外,257 例进行了相应基因测序,基因芯片检测结果经测序复核为一致,在基因芯片检测结果的基础上,测出GJB2 c.235delC/c.109A>G 1例,SLC26A4 IVS7-2A>G/c.1173C>A 1例,GJB2c.235delC/SLC26A4IVS7-2A>G/SLC26A4 c.109G>T 1 例,其中GJB2 c.109A>G、SLC26A4c.1173C>A、SLC26A4 c.109G>T为芯片无法检测位点,经OMIM、HGMD 数据库查询均为已知致病突变。见图2。
2.3 新生儿听力筛查联合耳聋基因检测结果分析在进行了耳聋基因与听力筛查联合分析的6 957 例新生儿中,初筛未通过504 例,初筛阳性率7.24%。初筛未通过者在29~42 d 时接受复筛,复筛未通过88 例,复筛阳性率1.26%。听力筛查未通过者中88 例均在3 个月龄进行了听力学诊断,最终确诊5 例耳聋患者,分别为双耳极重度感音神经性耳聋2 例,左耳极重度感音神经性耳聋/右耳中重度感音神经性耳聋1 例、双耳中重度感音神经性耳聋1 例,2 例双耳极重度感应神经性耳聋患儿分别为GJB2 c.235delC 纯合突变,GJB2c.235delC/c.299_300delAT,左耳极重度感音神经性耳聋/右耳中重度感音神经性耳聋基因型为SLC26A4IVS7-2A>G/c.1173C>A,双耳中至重度感音神经性耳聋基因型为GJB2c.235delC/SLC26A4IVS7-2A>G/SLC26A4c.109G>T,双耳轻中度感音神经性耳聋1 例,基因型为GJB2 c.235delC/c.109 A>G。
3 讨论
自1988年第一个耳聋基因被定位,1995年发现第1 个NSHL 基因POU3F4 并被成功克隆[8],1997年发现最常见的NSHL致聋基因GJB2以来[9],20年来遗传性耳聋的研究取得了飞速发展。目前大量研究[10-15]证明,GJB2、SLC26A4、mtDNA12SrRNA是导致大部分NSHL 的主要致病基因。新生儿听力筛查联合基因检测在耳聋出生缺陷防控中具有重要意义。Joint Committee on Infant Hearing[3]、王秋菊等[6]分别在2006年、2007年提出了新生儿听力与基因联合筛查的理念,至今,联合筛查已在国内外广泛展开。

表1 部分测序引物序列Tab.1 Primer sequences of Sanger sequencing method
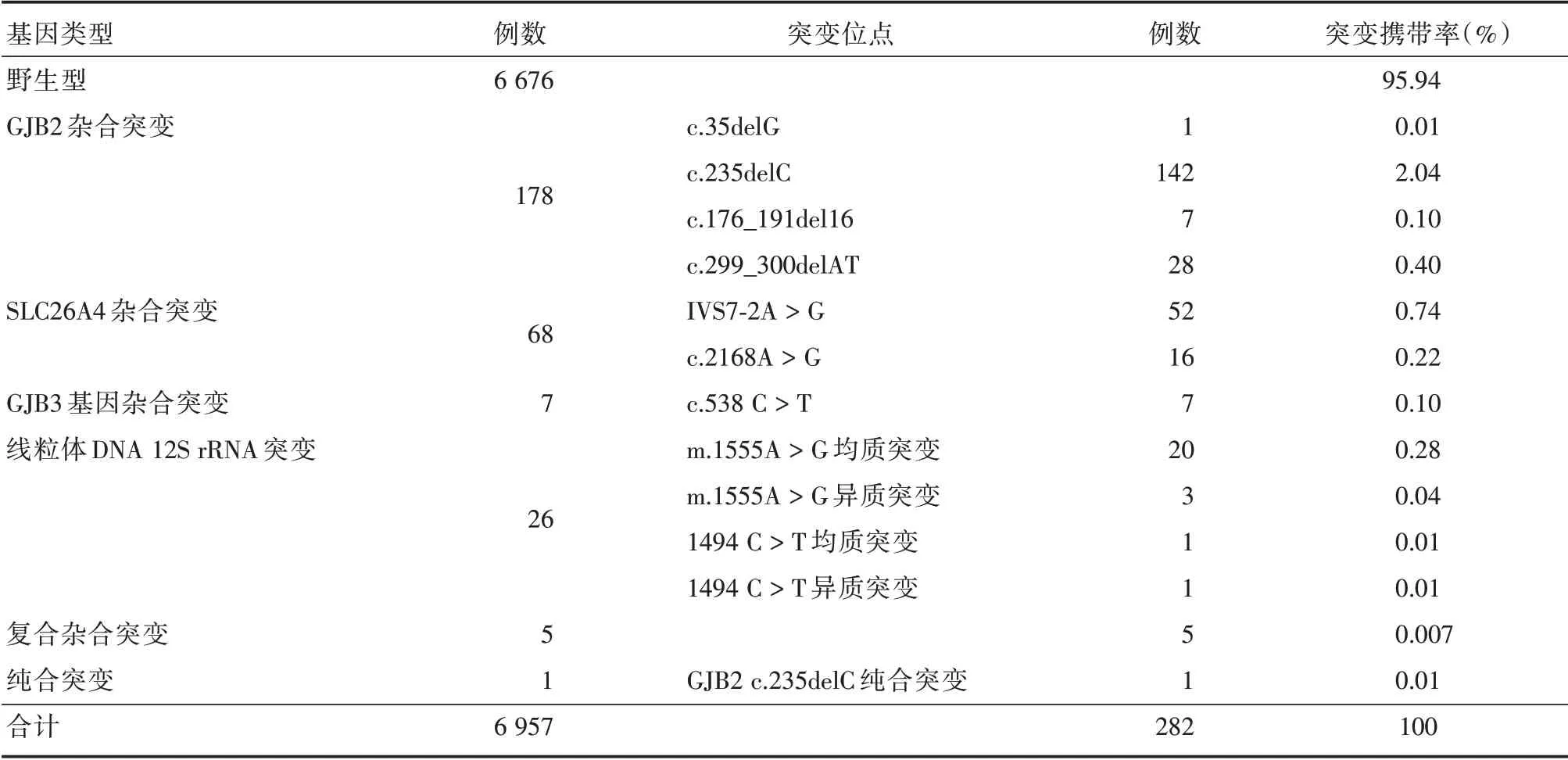
表2 6 957 例新生儿耳聋基因芯片结合基因测序检测结果突变类型及等位基因频率分布Tab.2 Mutation type and allele frequency of 6 957 cases of newborns detected by neonatal deafness gene chip combined with gene sequencing
6 957 例新生儿中,初筛未通过504 例,初筛阳性率7.24%。为了排除羊水或胎脂堵塞耳道、鼓室内有羊水、耵聍堵塞、外耳道狭窄、新生儿候鸣声干扰、机器校准有异常等因素对听力筛查的影响,初筛未通过者在29~42 d 时接受复筛,复筛未通过88 例,复筛阳性率1.26%,与国内多项研究[16-23]中新生儿听力筛查结果数据接近。

图1 基因芯片检测常见耳聋基因9 个突变位点原始数据图Fig.1 Original graph of 9 mutation sites of common deafness genes detected by gene chip
耳聋基因突变存在地域和种族差异,因此,明确当地耳聋基因突变谱对耳聋出生缺陷防控具有重要意义。江西地区人口流动率较低,本研究纳入的新生儿父母均为江西籍贯,本研究数据能反映本地区耳聋基因突变热点。在6 957 例新生儿中,检出携带者282 例,约占总数的4.05%,该数据接近于文献报道的4.17%~5.15%[16-23],共检出GJB2基因突变183 例,突变携带率为2.6%,其中GJB2 c.235delC 杂合突变携带142 例,GJB2 c.235delC 纯合突变1 例,GJB2c.235delC/c.299_300delAT 双杂合1 例、GJB2 c.235delC/c.109 A>G 双杂合1 例、GJB2c.235delC/m.1555A>G 均质突变1 例、GJB2 c.235delC/SLC26A4 IVS7-2A>G/SLC26A4 c.109G>T杂合突变1例、c.35delG杂合突变1例、GJB2c.176_191del16 杂合突变7 例、GJB2c.299_300 del AT 杂合突变28 例,GJB2 c.235delC 位点突变共147 例,突变携带率为2.1%,高于文献报道的1.651%~1.975%[16-23],GJB2 基因为本地区最常见的耳聋基因,其突变热点为GJB2 c.235delC,与上述文献结论一致。

图2 部分突变位点测序峰图Fig.2 Original graph of sequencing of some mutation sites
在6 957 例新生儿中,共检出SLC26A4 突变70例,其中IVS7-2A>G杂合突变52例,复合杂合突变2 例,分别为SLC26A4 IVS7-2A>G 与c.1173C>A 复合杂合突变、GJB2 c.235delC/SLC26A4 IVS7-2A>G/SLC26A4 c.109G>T杂合突变1例,SLC26A4基因突变携带率为1.006%,SLC26A4IVS7-2A>G突变携带率为0.77%,SLC26A4c.2168 A>G 杂合突变16 例,突变携带率为0.22%,与上述几个大样本研究数据接近。SLC26A4 为本地区第2 大耳聋易感基因,其突变热点为SLC26A4 IVS7-2A>G。
本研究共检测出GJB3 c.538C>T杂合突变7例,突变携带率为0.1%,低于文献[16-23]报道的0.324%~0.369%。检出线粒体突变共27 例,突变携带率为0.38%,均高于上述研究中的0.224%~0.345%,说明江西地区线粒体突变携带率较高,听力筛查联合耳聋易感基因检测极大的降低了本地区药物性耳聋发生率,具有重大意义。
对比以往研究,本研究特色在于对基因芯片检出耳聋基因携带者进行了相应基因测序,从而检出3 例复合杂合突变携带者,分别为GJB2 c.235delC/109 A>G
复合杂合突变,GJB2c.235delC/SLC26A4 IVS7-2A>G/SLC26A4 c.109G>T复合杂合突变,SLC26A4 IVS7-2A/c.1173C>A 复合杂合突变,此3 例均未通过两次听力筛查,并通过听性脑干反应确诊为先天性感音神经性耳聋,其中GJB2 c.235delC/109A>G突变为轻至中度耳聋,其余2 例均为重度感音神经性耳聋。
本次检测中测出的GJB2 c.109 A>G 的致病性尚存在一定争议,RYDZANICZ 等[24]研究表明其与轻至中度耳聋有关。
但越来越多的研究表明,GJB2 c.109 A>G 可能为良性多态。因此GJB2 c.235delC/109A>G 突变为轻至中度耳聋此例患者需进一步采用耳聋panel 或家系全外显子组测序技术对耳聋病因进行鉴定,笔者将持续随访并建议进一步检查,观测其听力损失状态。新生儿听力筛查联合耳聋基因检测对本地区耳聋三级防控具有重要意义,有利于发现迟发型耳聋、避免药物性耳聋,以及指导耳聋基因携带家系再生育,具有重大意义。
猜你喜欢
开卷有益·求医问药(2021年12期)2021-12-29 00:34:55
中医眼耳鼻喉杂志(2021年2期)2021-07-21 08:53:28
今日农业(2021年4期)2021-06-09 06:59:56
种子(2021年3期)2021-04-12 01:42:22
快乐语文(2018年31期)2018-03-01 11:22:56
现代检验医学杂志(2016年4期)2016-11-15 02:01:00
外语教学理论与实践(2016年1期)2016-06-11 05:51:48
哈尔滨医药(2015年3期)2015-12-01 03:57:44
应用数学与计算数学学报(2014年2期)2014-09-26 05:40:23
中国CT和MRI杂志(2014年7期)2014-06-27 05:49:13
