
HPLC-MS/MS法鉴别中成药和保健食品中的双羟萘酸噻嘧啶
2020-07-24 02:07:56杨丽蓉聂晓华
临床医药文献杂志(电子版) 2020年38期
杨丽蓉*,张 筱,聂晓华,王 悦
(潍坊市检验检测中心,山东 潍坊 261200)
双羟萘酸噻嘧啶为淡黄色粉末,无臭,无味。易溶于碱溶液,在N,N-二甲基甲酰胺中略溶,在乙醇中极微溶解,在水中几乎不溶[1]。双羟萘酸噻嘧啶的检测方法收载在《中国药典》2015年版二部中,其主要为化学反应的理化鉴别法、高效液相色谱法、紫外分光光度法和红外分光光度法等。近年来在中成药和保健食品中非法添加化学药品的情况屡屡发生[2-5]。近期又发现有在中成药和保健食品中添加双羟萘酸噻嘧啶的情况,且与其相关文献报道比较少。本文建立HPLC-MS/MS的方法鉴别中成药和保健食品中添加的双羟萘酸噻嘧啶。
1 实验材料
1.1 实验样品
驱虫巧克力,批号(B)P51571
1.2 实验仪器
岛津LC-20A液相色谱串联AB4000+三重四级杆质谱仪
1.3 实验用对照品
双羟萘酸噻嘧啶对照品纯度:99.0﹪。
1.4 试剂
乙腈,乙酸,氨水,乙酸铵,试验用水为超纯水
2 检测方法与结果
2.1 液相色谱条件
a)色谱柱:C18柱,5μm,4.6×150 mm。b)流动相:乙腈:20 mmol/L乙酸铵(80:20)。c)流速:0.3 ml/min。d)进样量:1.0 μL 。
2.2 质谱检测条件
1)电离方式:电喷雾正负离子模式。2)检测方式:多反应监测模式(MRM)。3)雾化气:高纯氮气。4)气帘气:10psi。5)喷雾气:13psi。6)离子喷雾电压:4500(+,-)。7)主要质谱参数见表1。
2.3 对照品溶液的制备
精密称取双羟萘酸噻嘧啶对照品约0.025 g,至25 ml量瓶中,用溶液A[乙腈:水:乙酸:氨水(93.4:3:3:0.6)]溶解并稀释至刻度,再精密量取1.00 ml至100 ml量瓶中,用甲醇稀释至刻度。用微孔滤膜过滤(0.22 μm,下同)备用。
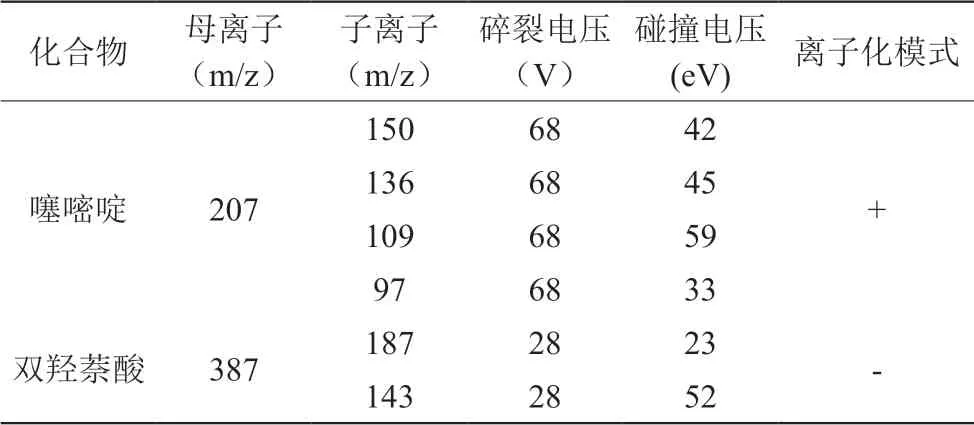
表1 质谱参数
2.4 供试品溶液的制备
精密称取样品约0.5 g,置50 ml容量瓶中,加入溶液A适量,超声提取30min,放冷至室温,用溶液定容。精密量取1.00 ml至100 ml量瓶中,用甲醇定容,过滤,取续滤液作为待测液。
2.5 检出限
根据双羟萘酸噻嘧啶的S/N=3时溶液的溶度计算方法的检出限,检出限测定液的配置见2.5.1-2.5.3,各种剂型的检出限见表2。
2.5.1 称取六种基质各约2g,分别置100 ml量瓶中,用溶液A溶解并稀释至刻度,作为基质溶液。
2.5.2 称取双羟萘酸噻嘧啶对照品约25 mg置25 ml量瓶中,用溶液A溶解并稀释至刻度,再精密量取1.00 ml置100 ml量瓶中,用溶液稀释至刻度,得到10 μg/ml的对照品溶液。
2.5.3 以基质溶液为稀释剂,把10 μg/ml的对照品溶液逐级稀释,分别在噻嘧啶和双羟萘酸的S/N接近3时,确定检出限。
2.6 定性判定[6]
根据样品中色谱峰的保留时间与对照品中色谱峰的保留时间一致,以及样品中色谱峰定性离子对的相对丰度与浓度相当的对照品相对丰度一致进行判定。

表2 不同剂型的检出限(μg/g)
2.7 样品的测定
按照2.4项下方法,精密称取样品约0.5 g,置50 ml容量瓶中,加溶液A适量,超声提取30 min,放冷至室温,用溶液定容。精密量取1.00 ml至100 ml量瓶中,用甲醇定容,过滤,取续滤液作为待测液。样品中检测出双羟萘酸噻嘧啶。
2.8 样品相对离子丰度
样品相对离子丰度符合定性判定的要求,样品相对离子丰度见表3。

表3 样品相对离子丰度
3 讨 论
《中国药典》2015年版二部收载的双羟萘酸噻嘧啶的液相色谱鉴别方法,采用的是硅胶柱。硅胶柱为正向色谱柱,在药品检验中使用反向色谱柱较多,而使用正向色谱柱较少。正向色谱柱使用时要用正己烷、乙酸乙酯等过度,使用时较为复杂。且《中国药典》2015年版二部收载的双羟萘酸噻嘧啶的液相色谱鉴别方法,需要使用双羟萘酸对照品和双羟萘酸噻嘧啶对照品两种对照品,分别进行检测,通过两种对照品的保留时间一致进行定性判定,检测过程较为繁琐。采用HPLC-MS/MS法对双羟萘酸噻嘧啶进行鉴别,样品的测定在10min之内完成,既可以通过保留时间的一致性进行判定,又可以通过特征离子对进行判定,检测方法简便快捷。因此,也建议《中国药典》中增加液相-质谱联用多反应检测的方式对化合物进行定性鉴别。
猜你喜欢
化工设计通讯(2022年10期)2022-12-31 20:42:50
波谱学杂志(2022年2期)2022-06-14 09:52:02
昆明医科大学学报(2021年8期)2021-08-13 08:59:04
武警医学(2018年10期)2018-11-06 07:04:34
山东化工(2018年15期)2018-09-20 08:55:34
当代化工研究(2016年6期)2016-03-20 16:21:42
首都食品与医药(2015年18期)2015-11-03 05:59:08
现代检验医学杂志(2015年1期)2015-02-06 01:59:14
吉林地质(2014年4期)2014-03-11 16:47:54
无机化学学报(2014年3期)2014-02-28 17:30:55
