
手性1,2-氨基醇合成进展
2020-07-23 14:49季少天
辽宁化工 2020年7期
季少天
手性1,2-氨基醇合成进展
季少天
(温州大学 化学与材料工程学院,浙江 温州 325035)
手性1, 2-氨基醇作为合成砌块常被作为起始原料用于生物活性化合物或手性配体的制备,也可以作为手性辅基被直接用于不对称合成。本文简述了手性氨基醇的主要合成方法,并对近些年合成方法的进展予以整理报道。
氨基醇; 手性配体; 不对称催化
手性氨基醇片段常见于具有生物活性的天然产物或药物分子中[1-3],经常作为手性控制片段被应用在催化剂设计中,也常作为手性配体的起始原料应用于不对称催化。然而,目前常见的手性氨基醇往往来源于天然氨基酸的还原氢化,底物多样性非常受限,而手性非天然氨基酸的制备常用到剧毒的金属氰化物,且对映体选择性往往不尽人意。所以,开发高效手性氨基醇的合成方法无论是对药物研发还是手性配体发展都有重要意义。本文总结了常见的手性氨基醇制备方法,并对近些年的合成进展予以整理报道。
1 合成研究
目前常用的制备手性1,2-氨基醇的方法有:氨基酸还原法,烯烃双官能团化,环氧化合物开环氨化,手性辅基,曼尼希型反应。
1.1 氨基酸还原法
作为制备手性氨基醇最早的方法之一,氨基酸还原法无论在实验室制备还是在大规模工业生产中都有成熟的路线,但受限于天然氨基酸种类较少,光学纯非天然氨基酸合成困难,该方法实际可制备的氨基醇种类较少。
1992年,Atsushi Abiko[4]课题组报道了NaBH4- H2SO4体系将氨基酸还原至对应的氨基醇,该方法操作简单,原料廉价易得,并且可大量制备(Scheme 1)。

Scheme 1
1993年,McKennon[5]课题组报道了NaBH4-I2体系还原氨基酸,以THF为溶剂,0 ℃下加原料,当量碘单质,2.5当量硼氢化钠,回流搅拌得对应氨基醇(Scheme 2)。此外,该方法对于N-酰基保护氨基酸该体系可以以良好的收率同时得到还原羧基与N-酰基的产物。
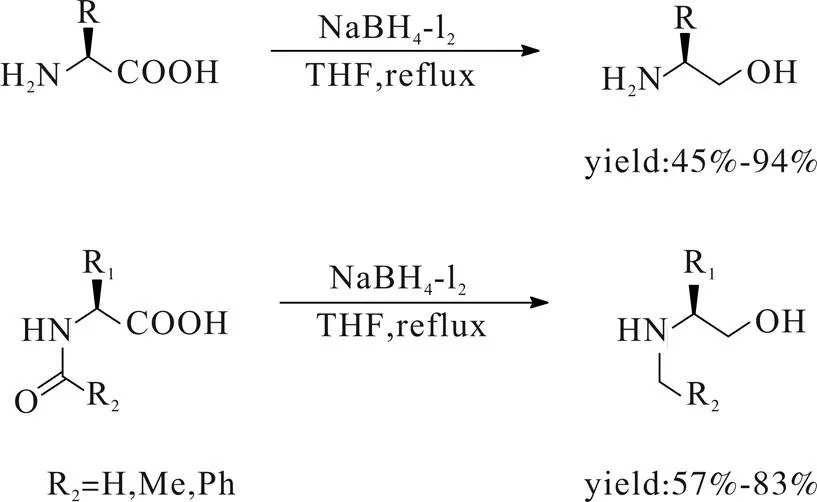
Scheme 2
1.2 烯烃双官能团化
通过烯烃双官能团化制备手性氨基醇是近些年来的研究热点,其优点在于烯烃底物种类繁多,来源广泛,可以通过该方法快速大量制备多取代手性氨基醇类化合物,极大扩充了氨基醇化合物的种类。2000年,K.Barry Sharpless[6]课题组报道了N-溴代酰胺为氮源的烯烃双官能团化构建手性氨基醇的方法。该方法以金属锇与(DHQ)2PHAL为催化剂,N-溴代酰胺为氮源,通过烯烃的双官能团化构建出一系列N-乙酰基氨基醇类化合物(Scheme 3)。和报道较多的N-氯代酰胺相比,N-溴代酰胺不易发生霍夫曼消除等副反应,可以通过反应温度调控获得更好的收率,而原料可使用DBI为溴化剂快速高效构建。该反应底物不仅限于末端烯烃,且对于脂肪族烯烃具有更好的区域选择性和立体选择性。

Scheme 3
2008年,Sherry R.Chemler[7]课题组报道了铜催化下分子内烯烃双官能团化反应,以噁唑啉类配体进行立体调控构建出一系列手性吡咯烷及吲哚类化合物(Scheme 4)。
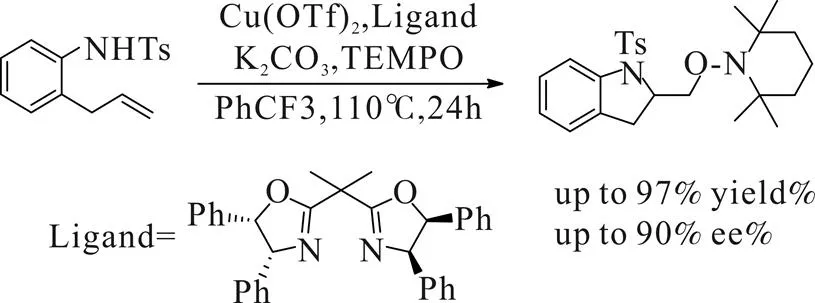
Scheme 4
2009年,Tehshi P.Yoon[8-9]课题组发展了一种铜催化下烯烃不对称双官能团化构建手性氨基醇的方法。该反应以烯烃与氧杂吖丙啶为原料,金属铜与手性噁唑啉配体为催化剂高效合成立体中心与氨基相连的手性四氢噁唑环,在盐酸条件下处理可方便获得对应的氨基醇类化合物。2012年,该课题组对反应的区域选择性进行调控,以金属铁与手性噁唑啉配体为催化剂高效合成立体中心与羟基相连的手性四氢噁唑环,同样在盐酸条件下处理可方便获得对应的氨基醇类化合物。该方法对芳香族末端烯烃具有较好的适用性,而对于脂肪族或非末端烯烃则不反应(Scheme 5)。

Scheme 5
1.3 环氧化合物开环氨化
环氧化合物开环氨化是制备手性氨基醇的经典方法之一。Salen配体的应用使环状环氧化合物的开环氨化取得非常优异的结果,近些年来随着对底物的研究,非环状环氧化合物的开环氨化也取得了极大进展,无论是末端无取代底物还是非末端环氧化合物也都能以合适的条件获得良好结果。然而该类反应仅对Salen-Cr(III)体系具有较好的选择性,其他手性催化体系对该类反应的研究仍有较大空间。
1997年,Jacobsen[11]课题组报道了合成环状手性氨基醇的方法,该方法以脂肪族杂环环氧化合物为原料,在三价铬与手性Salen配体配合物催化下与TMSN3发生开环反应,再经过后续处理获得对应手性氨基醇类化合物(Scheme 6)。然而,叠氮化合物具有潜在的危险性需要特殊的操作规程,且后续处理需要用到昂贵金属进行催化氢化,成本较高,步骤繁琐,操作条件不便,所以该方法并不能满足工业上大规模生产。
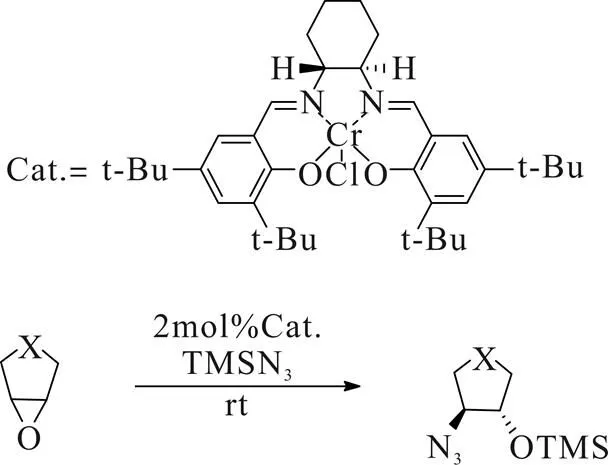
Scheme 6
2004年,Giuseppen Bartoli[12]课题组第一次报道了Boc氨对末端环氧化合物的氨解动力学拆分反应(Scheme 7)。该反应以外消旋的末端环氧化合物与Boc氨为原料,TBME为溶剂,手性Salen-Co(II)为催化剂,催化对硝基苯甲酸为氧化剂室温反应可获得良好的产率与优异的光学纯度。该方法所用催化剂方便易得,反应条件绿色环保,脱保护处理简便,不仅对脂肪族环氧化合物具有良好的选择性,对于一些因为电子效应与立体效应导致难以开环或者难以获得良好对映体选择性的芳香族化合物也具有良好的适用性。该方法缺点在于其反应本身为动力学拆分过程,底物利用率不高,而环氧化合物仅限于末端较为受限,未见放大制备。
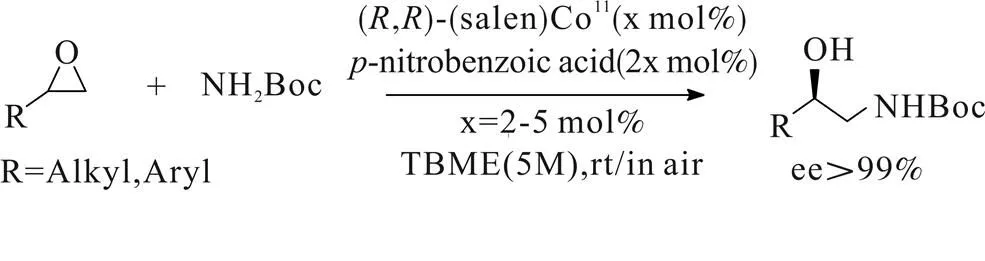
Scheme 7
2005年,Xavier Verdaguer[13]课题组报道了由手性环氧化合物开环氨化制备2-氨基-2-芳基-1,1-二芳基氨基醇的方法。该方法以手性环氧化合物为原料,2当量LiClO4为催化剂,10当量胺类化合物120~140 ℃无溶剂反应,得到中等至良好的产率以及优异的光学纯度,而对应的环氧化合物可用Jacobsen环氧化反应快速构建。该方法适用于不能由叠氮化合物开环制备的环氧底物(Scheme 8)。
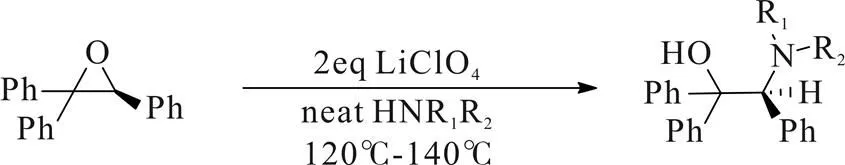
Scheme 8
2013年,Jacobsen[14]课题组报道了氨基甲酸苯酯对环状环氧化合物的加成反应,该反应以salen配体二聚物的钴配合物为手性催化剂,乙腈溶剂,50 ℃条件下反应48 h可得到中等至良好的收率以及优异的立体选择性(Scheme 9)。环状环氧化合物较末端环氧化合物反应活性更低,且由于酰胺类化合物本身亲核性不高,所以一般条件下难以反应。该方法使用的二聚体配合物作为催化剂相较于单独的Salen-Co(III)金属配合物,反应活性更高,立体选择性更好,使用量最低可降至0.2% (mol),且可以进行克级反应,为手性环状氨基醇的工业制备提供了一种强有力的手段。
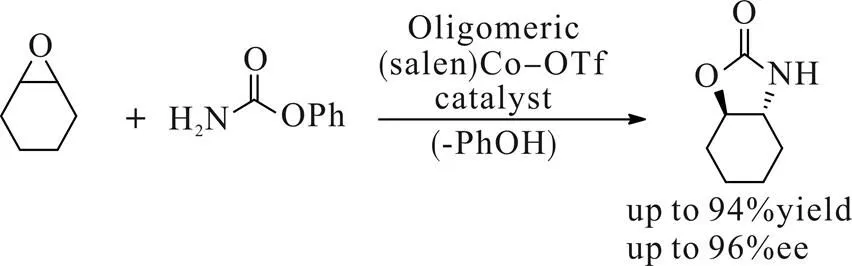
Scheme 9
1.4 手性辅基
2001年,Jonathan A.Ellman[15]课题组报道了由手性辅基制备β-取代氨基醇的方法,该方法以手性叔丁基亚磺酰醛亚胺或酮亚胺为原料,甲苯为溶剂,三甲基铝为Lewis酸催化剂,-78 ℃条件下与格氏试剂发生反应,再经过后处理脱除手性辅基得到对应的手性氨基醇(Scheme 10)。该方法对于脂肪族、芳香族或炔烃金属试剂都能得到优异的立体选择性。该反应路线可靠,底物普适性良好,后处理简单易行,所用手性源廉价易得,缺点在于需要脱除手性磺酰亚胺辅基,原子经济性不高。

Scheme 10
2011年,Jacob B.Schwarz[16]课题组通过氢化还原法制备了一系列芳基取代氨基醇类化合物(Scheme 11)。该方法以TBS保护的羟基芳基酮叔丁基磺酰亚胺为原料,金属氢化物或硼氢化物为氢源选择性还原,以良好的收率与优异的立体选择性获得氨基醇类化合物。该方法起始原料便于制备,氢源廉价易得,底物普适性较好,相较于有机金属化合物的曼尼希加成反应操作更为简便,条件也更温和经济。
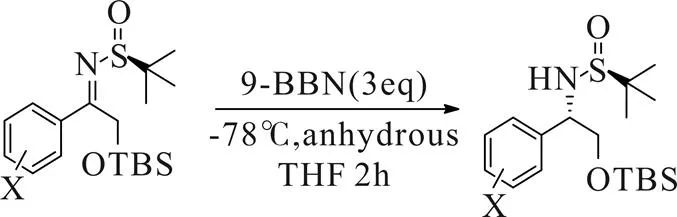
Scheme 11
2012年,Oscar Delgado[17]报道了由手性辅基制备β-芳基-β-烷基氨基醇的方法,该方法由硅基醚保护的α-羟基芳基酮和烷基金属试剂为原料,以手性叔丁基亚磺酰为手性辅基进行的曼尼希反应得到对应的氨基醇,dr值可达到97:3(Scheme 12)。

Scheme 12
1.5 曼尼希型反应
2002年,Harry J. Martin[18]课题组首次报道了不对称有机小分子催化三组分反应构建手性氨基醇的方法。该反应以廉价易得的()-脯氨酸为催化剂,α-羟基酮、醛和芳香胺为原料进行曼尼希加成,一锅法制备手性氨基醇类化合物。该反应系首次以有机小分子为催化剂进行的曼尼希反应,也是首次以游离醛参与反应的不对称催化曼尼希反应,具有重要意义。此外,氨基酸作为一种小分子催化剂,它的廉价易得与优异的立体选择性也彰显出其在不对称催化领域的广阔前景(Scheme 13)。

Scheme 13
2014年,林国强[19]课题组报道了一种由环状烷基酮磺酰亚胺芳基化构建烷基-芳基氨基醇的方法(Scheme 14)。该方法以过渡金属铑及手性二烯配体为催化剂,芳基硼烷对磺酰亚胺进行加成反应,以良好的收率与优异的立体选择性制备出一系列手性环状磺酰胺类化合物,再经过简单还原即可得到对应氨基醇类化合物。缺点是该方法仅对对位含供电子基芳硼酸有良好的反应性,含氯或羧基等吸电子基芳硼酸收率较低。
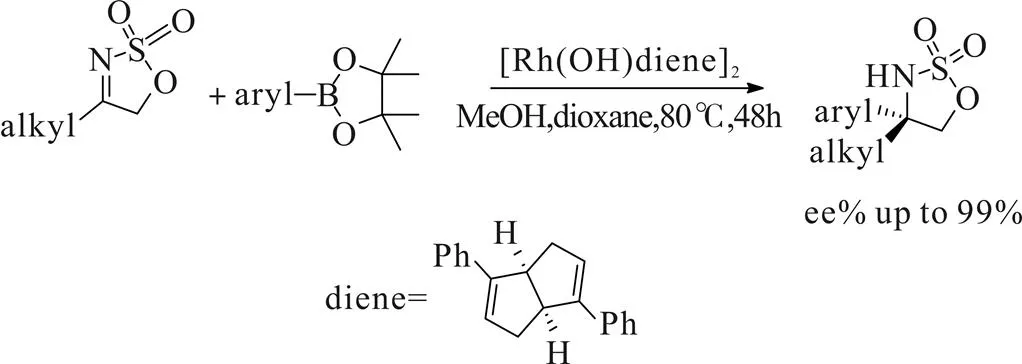
Scheme 14
2017年,徐明华[20]课题组报道了类似的反应,该反应以过渡金属铑及烯烃-手性亚磺酰胺配体为催化剂,同样良好的收率和优异的立体选择性获得烷基芳基氨基醇类化合物(Scheme 15)。该催化体系与铑手性二烯催化体系相比有更高的反应活性,并且手性配体的结构更易修饰改造,合成更简便快捷。该反应对含供电子基团的芳基硼酸具有较高的活性,对于含吸电子基团的芳基硼酸虽然活性较低,但可以通过提高硼酸当量获得较好产率。同时,对于由空间位阻导致难以反应的邻取代芳基硼酸也可以用该体系得到少量产物。

Scheme 15
2 结束语
随着近些年来合成化学的飞速发展,手性氨基醇类化合物的制备方法愈见多样。除上述较为常见的几种类型外,自由基型偶联反应[21]也偶有报道,然而适用性与对映体选择性还有诸多限制。而传统方法也具有需使用贵金属,手性催化剂价格昂贵以及反应放大困难等限制,所以新合成途径的开发以及传统方法的绿色合成研究仍然具有重要意义。
[1] STEPHEN C B. The synthesis of vicinal amino alcohols[J]., 2000, 56 (17):2561-2576.
[2] LAI Y S,MENDOZA J S,JAGDMANN G E,et al. Synthesis and protein kinase C inhibitory activities of balanol analogs with replacement of the perhydroazepine moiety[J].,1997,40(2):226-235.
[3] AGER D J,PRAKASH I,SCHAAD D R. 1,2-Amino alcohols and their heterocyclic derivatives as chiral auxiliaries in asymmetric synthesis[J].,1996,96(2):835–876.
[4] ABIKO A,MASAMUNE S. An improved, convenient procedure for reduction of amino acids to aminoalcohols: Use of NaBH4-H2SO4[J].,1992,33(38):5517-5518.
[5] MCKENNONAND M J, MEYERS A I. A convenient reduction of amino acids and their derivatives[J].1993,58(13):3568-3571.
[6] DEMKO Z P,BARTSCH M,SHARPLESS K B. Primary amides,a general nitrogen source for catalytic asymmetric aminohydroxylation of olefins[J].,2000,2(15):2221-2223.
[7] PETER H F, JIN-WOO K,SHERRY R.Copper catalyzed enantio- selective intramolecular aminooxygenation of alkenes [J].,2008,130(52):17638-17639.
[8] DAVID J M, KEVIN S W, TEHSHIK P Y. Oxaziridine-mediated enantioselective aminohydroxylation of styrenes catalyzed by copper(II) bis(oxazoline) complexes[J].,2009,65(26):5118-5124.
[9] KEVIN S. WILLIAMSON,TEHSHIK P. Iron catalyzed asymmetric oxyamination of olefins[J]., 2012,134(30):12370-12373.
[10] SCHAUS S E, LARROW J F, JACOBSEN E N. Practical synthesis of enantiopure cyclic 1,2-amino alcohols via catalytic asymmetric ring opening of meso epoxides[J]., 1997, 62(12):4197-4199.
[11] BARTOLI G, BOSCO M, SAMBRI L. Asymmetric catalytic synthesis of enantiopure n-protected 1,2-amino alcohols[J].,2004, 6(22):3973-3978.
[12] GARCIA-DELGADO N, REDDY K S, VERDAGUER X. Synthesis of heavily substitutes 1,2-amino alcohols in enantiomerically pure form[J].,2005,70(18):7426-7433.
[13] BIRRELL J A, JACOBSEN E N. A practical method for the synthesis of enantioenriched trans-1,2-amino alcohols[J].,2013, 15(12):2895–2897.
[14] TANG T P,VOLKMAN S K,ELLMAN J A. Asymmetric synthesis of protected 1,2-amino alcohols using tert-butanesulfinyl aldimines and ketimines[J]., 2001, 66 (26):8772–8778.
[15] PAN X G, JIA L B, LIU X j,et al. A general asymmetric synthesis of phenylglycinols[J].,2011,22(3):329-337.
[16] DELGADO O, MONTEAGUDO A, FUSTERO S. A practical entry to β-aryl- β-alkyl amino alcohols:application to the synthesis of a potent BACE1 inhibitor[J].,2012, 10(33):6758-6766.
[17] LIST B, POJARLIEV P, BILLER W T, et al. The proline-catalyzed direct asymmetric three-component Mannich reaction: scope, optimization, and application to the highly enantioselective synthesis of 1,2-amino alcohols[J].,2002, 124(5):827-833.
[18] CHEN Y J, CHEN Y H, FENG C G , et al. Enantioselective rhodium- catalyzed arylation og cyclic n-sulfamidate alkylketimines: a new access to chiral β-alkyl-β-aryl amino alcohols[J].,2014,16(12):3400–3403.
[19] LIU M Q, JIANG T, CHEN W W, et al. Highly enantioselective Rh/chiral sulfur-olefin-catalyzed arylation of alkyl-substituted non-benzofused cyclic N-sulfonyl ketimines[J].,2017,4(11):2159-2162.
[20] DING W, LU L Q, XIAO W J. Visible light photocatalytic radical- radical cross-coupling reactions of amines and carbonyls:a route to 1,2-amino alcohols[J]., 2016, 81 (16):7237–7243.
Progress of Synthesis of Chiral 1,2-Amino Alcohol
(School of Chemistry and Material engineering, Wenzhou University, Wenzhou Zhejiang 325035, China)
Chiral 1,2-amino alcohols is not only the important synthon in the synthesis of bioactive compounds and chiral ligands,but also chiral cogroups for asymmetric synthesis.In this paper, the main synthetic methods of chiral amino alcohols were described,and the progress of new methods in recent years was discussed.
Chiral 1,2-amino alcohols ; Chiral ligands; Asymmetric synthesis
2020-02-28
季少天(1994-),男,硕士,安徽阜阳人,研究方向:不对称催化。
TQ 201
A
1004-0935(2020)07-0874-05
猜你喜欢
大电机技术(2022年5期)2022-11-17
中南民族大学学报(自然科学版)(2022年6期)2022-11-02
分子催化(2022年1期)2022-11-02
功能材料(2022年5期)2022-06-02
中南民族大学学报(自然科学版)(2020年6期)2020-12-22
山西化工(2020年2期)2020-02-16
世界农药(2019年4期)2019-12-30
筑路机械与施工机械化(2017年6期)2017-07-10
航天制造技术(2016年6期)2016-05-09
中国塑料(2015年10期)2015-10-14
