
p型掺杂剂Cp2Mg在MOCVD气相中的反应机理研究
2020-07-21 07:15张红唐留
化工学报 2020年7期
张红,唐留
(江苏大学能源与动力工程学院,江苏镇江212013)
引 言
Ⅲ族氮化物,特别是纤锌矿结构(六方相)的AlN、GaN 和InN 及 其 合 金AlGaN、GaInN 和AlGaInN,由于它们都是直接带隙结构,禁带宽度从1.9 eV 至6.2 eV 连续可调,是制作可见光至近紫外光光电子器件(发光二极管、激光二极管等)的优良材料。金属有机化学气相沉积(MOCVD)是生长氮化物薄膜最广泛采用的方法。在MOCVD 过程中,通常以Ⅲ族源气体、掺杂剂和NH3为前体通入反应器,在高温衬底上发生化学反应,生长单晶薄膜。尽管MOCVD 是一种非常成功的生长氮化物材料的方法,但由于生长中复杂的气相寄生反应[1-3],造成源气体利用率低[2-4]、表面形貌差[5-7]和p 型掺杂困难[8-9]等问题。
在早期GaN 的研究中,通过掺Zn 进行p 型掺杂,但效果并不明显,随后转向研究掺Mg 实现p 型掺杂。经实验测量和理论计算发现,Mg在GaN中引入的是深受主能级,最浅的受主能级在价带边之上170 MeV(1 MeV=1.6021766208×10⁻¹³J)[10]。而Mg 原子在GaAs 中引入的能级深度仅28 MeV。通常,室温下GaN 中仅有1%~2%的Mg 原子离化起受主的作用。因此,要获得高空穴载流子浓度,应该掺入比设计空穴浓度高约两个数量级浓度的Mg 原子。然而,高掺入Mg 原子并不一定能获得高空穴浓度,这是由于Mg 原子在生长过程中能够产生一些络合物,如与有机源和载气中的H 自由基形成Mg-H 络合物等,络合物对Mg 原子起钝化作用使Mg 原子难以激活离化[11]。另外,高Mg源流量会严重影响外延薄膜的表面形貌,甚至会使薄膜产生反相畴(inversion domain),改变薄膜材料的极性。大量的实验也表明,如果要使Mg 形成受主,需要将Mg-H 键断 开[12-20]。 最 早 取 得 成 功 的 是Amano 等[21]和Nakamura 等[22],他们分别利用低能电子辐照(LEEBI)技术和快速热退火(RTA)方法处理Mg掺杂GaN并成功合成低阻p 型GaN 材料,这是GaN 材料历史发展的一大里程碑。上述工艺特别适合工业化生产,是目前p 型GaN 实现Mg 受主激活的标准手段。尽管对于GaN 或InGaN 的p 型掺杂都取得了较好的效果,但是对于AlGaN,Mg 原子在AlxGa1-xN 中的能级更深[23],因此高Al 组分AlGaN 的p 型掺杂仍是目前的难题。Zvanut 等[24]通过测量与受体相关的电子顺磁共振(EPR)信号强度,研究了AlGaN∶Mg薄膜中H、N 和Mg 的相互作用,发现氢气钝化和激活Mg 的速率取决于AlxGa1-xN 层中Al 的组分。关于对AlGaN进 行p 型 掺 杂 的 研 究 现 状,Keller 等[8]和Li 等[9]已 经做了很好的综述,本文不再赘述。对于薄膜中Mg与周围原子的结合情况,前人也有相关研究。Götz等[17]利用低温傅里叶变换红外吸收光谱研究了GaN∶Mg 的异质外延层,发现氢键合到氮原子上形成Mg-N-H 络合物而不是简单的Mg-H 络合物。Neugebauer 等[25]通过第一性原理计算也得出了类似的结论,此外还发现在GaN∶Mg固相中,中性的H 在Mg原子附近发生电离,而剩余的H 与最近邻N 原子键合。
目前,普遍认为Mg-H 络合物形成于外延层中[17,26-27]。但是MOCVD 气相过程中是否已经形成Mg-H 未见相关研究,气相过程中掺杂剂的反应机理研究相对较少。Haffouz 等[28]研究了GaN 生长时p型掺杂剂(MeCp)2Mg 的气相反应,通过激光散射实验发现(MeCp)2Mg 在气相中与NH3反应产生微小的固体颗粒,猜测这些固体颗粒极有可能是多聚物[(MeCp)Mg(NH2)]m(m≥2)。类似地,Wang 等[29]通过量子化学计算发现,Cp2Mg 在少量和过量的NH3氛围中分别会形成Cp2Mg∶NH3和Cp2Mg∶(NH3)2两种络合物。此外他们通过傅里叶变换红外线光谱实验(Fourier transform infrared spectroscopy, FTIR)发现,在常温下Cp2Mg 与NH3会发生寄生沉积。上述研究均与掺杂剂的气相加合反应相关,然而实际生长过程中,掺杂剂在气相中的反应很复杂,关于掺杂剂详细的气相反应机理及最终产物的研究目前未见相关报道。因此对于这一内容的补充研究对揭示p型掺杂机理有着重要意义。
针对前人研究的不足,本文对MOCVD 气相过程掺杂剂(Cp2Mg)的反应机理做了详细的理论计算。将证明Cp2Mg 有两条相互竞争的反应路径,加合路径和氢解路径。气相中的H 自由基有利有弊,虽然H 自由基会降低Cp2Mg 的分解温度,但也会对Mg 产生钝化作用。Cp2Mg 在气相中的最终有利产物是Mg-H 络合物。本文对Cp2Mg 加合路径和Cp2Mg 中第一个和第二个碳环的分解路径进行了详细研究,并结合实验现象对研究结论进行了合理性说明。
1 计算方法及验证
图1 所示的是Cp2Mg 在MOCVD 气相过程中的化学反应路径示意图,主要分为加合路径和氢解路径。加合路径(红色箭头所示)中,Cp2Mg 经过G1 和G2 连续与NH3发生络合反应。氢解路径(蓝色箭头所示)中,Cp2Mg 可能经过分子内反应G4 和G6 连续消去两个Cp 生成Mg 原子;由于高温气相中存在H自由基[30-31],所以Cp2Mg或中间产物也可能受到H自由基的攻击发生加合反应(如G3、G9、G10、G12 和G13),然后再发生分子内反应(如G5、G11 和G14)消去CpH。值得注意的是,H 自由基的攻击可能有两种情况:H自由基对Cp碳环的攻击和对Mg的攻击。比如,Cp2Mg 在H 自由基的攻击下就可能生成(CpH)CpMg 和Cp2MgH 两种产物。最后,也提出了气相络合物Mg-H的形成机理。

图1 Cp2Mg在MOCVD气相过程中的化学反应路径示意图,主要分为加合路径(红色箭头所示)和氢解路径(蓝色箭头所示),其中虚线表示理论上不可能实现的反应Fig.1 Schematic diagram of the chemical reaction pathway of Cp2Mg in the MOCVD gas-phase process,which is mainly composed of the adduct reaction path(shown by the red arrow)and the hydrogenolysis reaction path(shown by the blue arrow),where the dotted line indicates the theoretically impossible reactions
本文利用量子化学的密度泛函理论方法,在广义梯度近似(GGA)和密度泛函理论(DFT)的框架内,使用GAUSSIAN 09 程序包[32]进行计算。通过B3LYP 方法[33]和6-31G(d)基组,对反应物和过渡态进行结构优化和频率计算。通过对在势能表面上计算出的固定点进行简振模式分析,合理的优化结构不存在虚数频率。对于过渡态结构,只存在一个虚数频率。此外,对过渡态进一步进行内禀反应坐标(IRC)[34]计算,以验证所找到的过渡态能连接反应物和产物。值得注意的是,在高温下(尤其是温度超过1500 K 时),分子结构的扭振频率会降低,这可能会导致振动配分函数的非物理性增加,从而导致自由能的增加。虽然本文的计算温度并不高,但是也考虑了简谐近似造成的能量误差,谐振频率校正因子取值为0.97,使用开源算法KiSThelP (kinetic and statistical thermodynamical package)对各物质的热力学性质进行计算,具体能量误差的修正和计算细节可参考Canneaux等[35]的工作。
在MOCVD 反应器中,高温衬底上方存在大的温度梯度。因此计算了不同温度下(293.15 ~1473.15 K)各反应的Gibbs自由能差(ΔG),通过ΔG判断反应进行的方向,自发反应必须满足ΔG<0 且ΔG越小反应发生的趋势越大。对于存在过渡态的反应,进一步用活化自由能(ΔG≠)来判断反应速率的快慢,ΔG≠越小反应速率越大反之则越小。从热力学和动力学判断反应路径的可能性。
根据化学热力学,Gibbs 自由能G可以用式(1)计算[36]

其中,Eelec、Evib(0)、Evib、Erot和Etrans分别是电子能量、零点振动能、振动能、转动能和平动能;R是气体常数;n是气体含量;S是熵值。

图2 加合路径中主要物质的优化结构Fig.2 Optimized structures of the main species in adduct reaction path
图2 所示的是加合路径中主要物质的优化结构,Cp2Mg具有类似于“三明治”的单体的夹层结构,其中环戊二烯基环是平行相对,中间是Mg 原子。在气相和结晶相中碳环与镁原子η5 键合[37-39],表1[29,37,40-41]中列出了各物质结构的具体键长及与文献计算值或实验值的对比。对比发现本文计算的Cp2Mg 优化结构与电子衍射和X 射线衍射的实验结构非常吻合[37,40],与文献计算值也基本一致[29,41]。对于Cp2Mg:NH3和Cp2Mg:(NH3)2的优化结构也与文献计算值非常吻合。因此本文所采用的理论层级和基底是合理的。
2 结果与讨论
2.1 Cp2Mg与NH3的加合路径(G1和G2)

在MOCVD 生长Ⅲ族氮化物薄膜时,气相环境中通常会存在大量NH3,Cp2Mg可能会与NH3发生加合反应(G1 和G2)形成Cp2Mg∶NH3或Cp2Mg∶(NH3)2。Cp2Mg∶(NH3)2有可能在气源进口处的低温附近形成,而在高温腔体区域存在的可能性很小。

图3 不同温度下(T=293.15 ~1473.15 K)加合反应(G1和G2)的Gibbs自由能差(ΔG)Fig.3 Changes of Gibbs free energy(ΔG)of adduct reactions(G1 and G2)at different temperatures(T=293.15—1473.15 K)
2.2 Cp2Mg中的第一个碳环氢解过程(G3~G8)

Cp2Mg 中的第一个碳环氢解过程如反应G3~G8所示。Cp2Mg可能通过分子内分解反应(G4和G6)生成Mg,然后Mg原子可能与H自由基连续反应(G7和G8)生成MgH2。Cp2Mg 也可能发生氢解反应G3 和G5 生成CpMg。上文已提到碳环Cp 和Mg 都有可能受到H 自由基的攻击形成不同的加合物。Cp2Mg 中的Mg 受到H 自由基的攻击会生成Cp2MgH,Cp2Mg和Cp2MgH 的自由能列在表2 中。发现在所有计算温度下Cp2MgH的自由能相对于Cp2Mg都是增大的,因此从热力学角度分析Cp2MgH 不可能形成。所以,Cp2Mg与H自由基的反应产物为(CpH)CpMg。同样地,CpMgH 消去Cp 形成MgH 的反应在理论上也不可能发生。

表1 主要物质的优化键长和键角Table 1 Optimized bond lengths and angles of the main species
图4所示的是氢解路径中主要物质和过渡态的优化结构。大部分结构中Mg 与碳环之间的距离约为2 Å,碳环上的C—C 键长约为1.45 Å。对于图4(e)中(CpH)Mg 的结构,发现Mg 与CpH 碳环之间的距离较长约为3.968 Å。这可能是(CpH)Mg 中的CpH 碳环已经形成相对稳定的电子结构,使得CpH碳环和Mg 之间的相互作用力降低,最终导致Mg 与CpH 碳环之间的距离相对于其他结构变长。对于过渡态图4(a)和(g),C—H 键过渡态键长约为1.82 Å。对于过渡态图4(c),Mg 与CpH 的距离伸长了约65%达到3.280 Å。

表2 不同温度下的相对Gibbs自由能Table 2 Calculated relative Gibbs free energy at different temperatures

图4 氢解路径中主要物质和过渡态的优化结构Fig.4 Optimized structures of the main species and transition states in hydrogenolysis reaction path

图5 不同温度下(T=293.15 ~1473.15 K)氢解路径(G3~G8)的Gibbs自由能差(ΔG)Fig.5 Changes of Gibbs free energy(ΔG)of hydrogenolysis reactions(G3—G8)at different temperatures(T=293.15—1473.15 K)
图5 所示的是不同温度下G3~G8 的Gibbs 自由能差ΔG。类似地,通过线性插值得到G3、G4、G5 和G6 的平衡温度分别为:Te(G3)= 809 K、Te(G4)= 1368 K、Te(G5)=360 K 和Te(G6)=1019 K。对于G3,当T<809 K 时,Cp2Mg 与H 自由基可越过很低的能垒自发反应生成(CpH)CpMg;对于G5,当T>360 K 时,(CpH)CpMg也可越过很低的能垒自发消去CpH生成CpMg;对于G6,当T>1019 K 时,CpMg 才可自发反应消去Cp 生成Mg;同样地,对于G4, 当T>1368 K时,Cp2Mg 才可自发反应消去Cp 生成CpMg。通过分析各反应发生的有利温度区间可以发现,低温下有利于Cp2Mg 与H 自由基的加合反应,而高温下有利于分解反应。继续分析,由Cp2Mg到CpMg存在两条路径,第一条路径(G3→G5):Cp2Mg先与H 自由基加合,然后消去CpH 生成CpMg;第二条路径(G4):Cp2Mg 直接发生分子内反应生成CpMg。对比上述两条反应路径发现,第一条路径在很低的温度下(T>360 K)即可发生。而第二条路径只有在已经接近衬底的高温下(T>1368 K)才能发生,所以在气相环境的温度范围内,Cp2Mg 很难通过分子内反应直接生成CpMg。具体原因可能是H 自由基结合Cp碳环形成CpH 后,减弱了CpH 与Mg 原子间的作用力,所以(CpH)CpMg 在低温下便可消去CpH。但是Cp2Mg 中的Cp 碳环与Mg 原子间的作用力依然很强,所以Cp2Mg 很难消去Cp。因此,H 自由基对Cp2Mg 中第一个Cp碳环的消去有重要的辅助作用。对于G7和G8,在所有温度下均可自发进行,因此,可以推断一旦气相中生成Mg 原子,那么Mg 原子会很快与H 自由基结合生成Mg-H络合物。
2.3 Cp2Mg中的第二个碳环氢解过程(G9~G14)

Cp2Mg 中第二个碳环的消去过程如反应G9~G14 所示。图6 所示的是不同温度下反应G9~G14的Gibbs自由能差。类似地,估算得到G13、G14 和G12 的平衡温度分别为:Te(G13)= 849 K、Te(G14)= 366 K 和Te(G12)= 1240 K。对于G13,当T<849 K 时,CpMgH 与H 自由基可越过很低的能垒自发反应生成(CpH)MgH;对于G14,当T>366 K 时,(CpH)MgH 可自发消去CpH 生成MgH;对于G12,当T<1240 K 时,(CpH)Mg 与H 自由基可自发反应生成(CpH)MgH。对于G9 和G11, 在所有计算温度下都可以自发进行。CpMg生成后,CpMg中的Cp碳环和Mg 都有可能受到H 自由基的攻击。所以相应地,CpMg可能有以下三条分解路径,第一条路径(G10→G13→G14):H 自由基先攻击CpMg 中的Mg(G10)生成CpMgH,然后继续攻击CpMgH 中的Cp(G13)生成(CpH)MgH。最后(CpH)MgH 消去CpH(G14)生成MgH。第 二 条 路 径(G9→G11):H 自 由 基 先 攻 击CpMg 中的Cp(G9)生成(CpH)Mg,然后(CpH)Mg 消去CpH(G11)生成Mg。第三条路径是交叉路径(G9→G12→G14):反应G9 生成的(CpH)Mg 再与H 自由基继续反应(G12)生成(CpH)MgH,然后经过G14 生成MgH。分析发现,第一条路径在很低的温度下(T>366 K)便可自发进行,第二条路径及交叉路径在所有计算温度下几乎都可以自发进行。对比上述三条路径与2.2 节中CpMg 通过分子内直接生成Mg(G6)发现,G6 的自发进行需要很高的温度,而CpMg在H 自由基的作用下可以大大降低分解温度。因此,H 自由基对Cp2Mg 中第二个Cp 碳环的消去也有重要的辅助作用。
2.4 相关实验证明
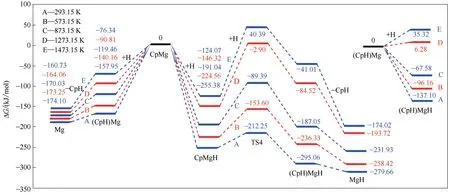
图6 不同温度下(T=293.15~1473.15 K)氢解路径(G9~G14)的Gibbs自由能差(ΔG)Fig.6 Changes of Gibbs free energy(ΔG)of hydrogenolysis reactions(G9—G14)at different temperatures(T=293.15—1473.15 K)
实际生长Ⅲ族氮化物通常都是处于高密封性和高温环境的MOCVD 反应腔中,所以对气相组分的原位测量非常困难。但是目前已有相关实验结果可以间接验证本文提出的Cp2Mg 气相反应机理。比如,对于加合反应路径,Wang 等[29]通过傅里叶变换红外线光谱实验发现,常温下,当Cp2Mg 和NH3通过分隔进口进入反应腔时,壁面没有观察到沉积;当二者混合进入时,壁面观察到朦胧的白色薄膜沉积。红外光谱数据表明,MgCp2和NH3的加合产物在不同的压力下相似但截然不同,低压氛围下为Cp2Mg∶NH3在高压生长氛围下为Cp2Mg∶(NH3)2。对于氢解路径的合理性,Nakamura 等[11]的实验结果能够有很好的验证。他们将在N2气氛下经过热退火或低能电子束辐照处理获得的低电阻率p型GaN 薄膜置于温度超过600℃的NH3氛围中再次高温退火后发现其电阻率高达16 Ω·cm。而在室温和1000℃之间的N2气氛中热退火的情况下,低电阻率p 型GaN薄膜的电阻率没有变化。他们认为这是由于有机源、载气或NH3分解产生的H自由基会形成Mg-H络合物使Mg原子难以激活离化。
根据对前人已有的实验现象和实验结果的分析,可以证明本文提出的反应机理符合实验现象。遗憾的是,H 自由基在辅助Cp2Mg 分解的同时也会对Mg 产生钝化作用。Cp2Mg 氢解反应虽然能在较低温度范围内进行,但是最终产物是MgH 或MgH2,这使得大部分Mg 不能够激活,空穴浓度下降,薄膜显示高阻性。因此,H 自由基的存在对Cp2Mg 而言是一把“双刃剑”。此外,本文只考虑了一个H 自由基参与反应的情况,实际上也可能有多个H 自由基或其他自由基参与反应,这仍需要进一步研究。本研究均是基于化学反应热力学和动力学,这是反应能够进行需要满足的前提。对于反应能进行到何种程度,各反应物质的实际浓度则需要结合流体动力学分析,将在下一步工作中继续研究。
3 结 论
本文对MOCVD 气相过程中p 型掺杂剂Cp2Mg的反应路径在不同温度下进行了量子化学计算。通过对比不同温度下不同反应路径上的Gibbs 自由能的变化,寻找Cp2Mg 的可能的路径和最终气相产物。根据对理论计算结果的分析,得到结论如下。
(1)Cp2Mg 主要有两条相互竞争的反应路径:加合路径和氢解路径。
(2)在加合路径中,293~573 K 的温度范围内,Cp2Mg 可能会与NH3通过1∶1(低氨氛围)或1∶2(高氨氛围)的比例加合,分别形成络合物Cp2Mg∶NH3和Cp2Mg∶(NH3)2。
(3)对于氢解路径,气相中的H 自由基会进攻Cp2Mg 或中间产物的碳环与Mg,从而减弱了Mg 与碳环之间的作用力,降低分解温度。尽管H 自由基能够促进Cp2Mg 的分解,但是最终形成的Mg-H 络合物对p型掺杂是极为不利的。
符 号 说 明
Eelec——电子势能,kJ·mol-1
Erot,,Etrans,Evib(0),Evib——分别为转动能、平动能、零点振动能和振动能,kJ·mol-1
G——Gibbs自由能,kJ·mol-1
ΔG——Gibbs自由能差,kJ·mol-1
ΔG≠——活化自由能,kJ·mol-1
n——气体含量,mol
R——气体常数,8.314 J·mol-1·K-1
S——熵值,kJ·mol-1·K-1
猜你喜欢
北京航空航天大学学报(2022年5期)2022-06-06
供水技术(2022年1期)2022-04-19
陶瓷学报(2021年4期)2021-10-14
保鲜与加工(2021年1期)2021-02-06
当代水产(2020年4期)2020-06-16
当代水产(2020年3期)2020-06-15
科学中国人(2018年8期)2018-07-23
电脑知识与技术(2018年3期)2018-03-21
哈尔滨理工大学学报(2017年1期)2017-04-08
科技视界(2016年24期)2016-10-11
