
RP-HPLC法测定甘氨酸原料药有关物质研究
2020-07-16 03:01赵士敏施细文
中国新技术新产品 2020年9期
赵士敏 许 岗 施细文 王 敏
(湖南福来格生物技术有限公司,湖南 长沙 410000)
甘氨酸(Glycine,缩写Gly)又名氨基乙酸,甘氨酸是内源性抗氧化剂还原性谷胱甘肽的组成氨基酸,机体发生严重应激时常用外源补充,有时也称为半必需氨基酸[1]。甘氨酸在食品、医药等领域应用广泛[2]。在医药行业中可以用作氨基酸制剂、抗胃酸冲剂以及其他赋形剂[3]。甘氨酸的工业生产方法主要有3 种,天然蛋白质水解法、氯乙酸氨解法和Strecker 法。目前国内普遍采用氯乙酸氨解法,国外则以Strecker 法为主。甘氨酸的原料药中可能会产生甘氨酸酐、亚氨基二乙酸、2-氯乙酸、双甘氨肽、三甘氨肽等杂质,但是国内尚未有关于测定甘氨酸原料药中这些杂质的报道。为了有效控制甘氨酸原料药的质量,保证用药安全,该实验采用HPLC法同时测定甘氨酸原料药中的上述5 种杂质,为原料药的质量控制提供参考。
1 试验部分
1.1 仪器与试剂
岛津LC-20AT 高效液相色谱仪;梅特勒320pH 酸度计;奥豪斯EX225DZH 十万分之一天平;抽滤装置、微孔滤膜(硝化纤维型,φ50 cm,孔径0.22 μm);上海一恒科学仪器有限公司[4]。
甘氨酸3 批:由湖南宝利士生物技术有限公司提供,甘氨酸酐(含量≥98%,标准物质),亚氨基二乙酸(含量≥98%,标准物质,),2-氯乙酸(含量≥98%,标准物质),双甘肽(含量≥98%,标准物质),三甘氨肽(含量≥98%,标准物质),以上标准物均有Sigma 公司提供。庚烷磺酸钠、磷酸(均为分析纯):天津科密欧化学试剂有限公司,超纯水:由DZG-303A 艾科超纯水机制得,乙腈(HPLC 级):Merck 公司[5]。
1.2 溶液的配制
流动相溶液:称取庚烷磺酸钠2.24 g,加水975 mL 溶解后,用磷酸调节pH 值至2.2,过滤后,再加入25 mL 乙腈,超声。
稀释剂溶液:同流动相溶液。
对照品储备溶液:称取各组分杂质对照品约25 mg,精密称定,置50 mL 量瓶中,用流动相溶解并稀释至刻度。(溶液浓度为500 μg/mL)。
各组分对照二级储备液:精密量取各组分对照品贮备液0.5 mL,置10 mL 量瓶中,加稀释剂稀释至刻度。(溶液浓度为25 μg/mL)。
对照品溶液:称取甘氨酸25 mg 和各组分对照品二级贮备液1.0 mL,置于同一50 mL 量瓶中,加稀释剂稀释至刻度。
各组分对照溶液:精密称取杂质甘氨酸酐、亚氨基二乙酸、2-氯乙酸、双甘氨肽、三甘氨肽对照品各约10 mg,分别至5 个50 mL 容量瓶中,加溶剂溶解后稀释至刻度,摇匀,分别作为各组分的对照溶液。(溶液浓度为0.5 mg/mL)。
供试品溶液:称取该品约25 mg,精密称定,置50 mL量瓶中,加稀释剂溶解并稀释至刻度。
80%限度加标供试品溶液:精密称取甘氨酸25.0 mg,加入50 mL 容量瓶中,加入各组分对照品二级贮备液0.8 mL,加稀释剂稀释至刻度,平行制备3 份。
100%限度加标供试品溶液:精密称取甘氨酸25.0 mg,加入50 mL 容量瓶中,加入各组分对照品二级贮备液1.0 mL,加稀释剂稀释至刻度,平行制备3 份。
120%限度加标供试品溶液:精密称取甘氨酸25.0 mg,加入50 mL 容量瓶中,加入各组分对照品二级贮备液1.2 mL,加稀释剂稀释至刻度,平行制备3 份。
1.3 分析方法
色谱柱CenturySIL C18-AQ(5 μm,4.6 mm×250 mm),流动相(含2.24 g/L 庚烷磺酸钠,用磷酸调节pH 值至2.2)-乙腈(97.5 ∶2.5),检测波长为200 nm,流速为1 mL/min,进样量20 μL,柱温25 ℃。
2 结果
2.1 系统适用性与仪器精密度
将对照品溶液连续进样6 针,考察各峰间的分离度、理论塔板数、拖尾因子、各峰保留时间的RSD、各峰峰面积的RSD。结果表明,在该条件下各峰间的分离度≥1.5,理论塔板数≥2 000,拖尾因子≤2.0。各峰保留时间RSD ≤1%,主峰面积RSD ≤2%,杂质峰面积RSD ≤10%。结果见表1,色谱图如图1 所示。
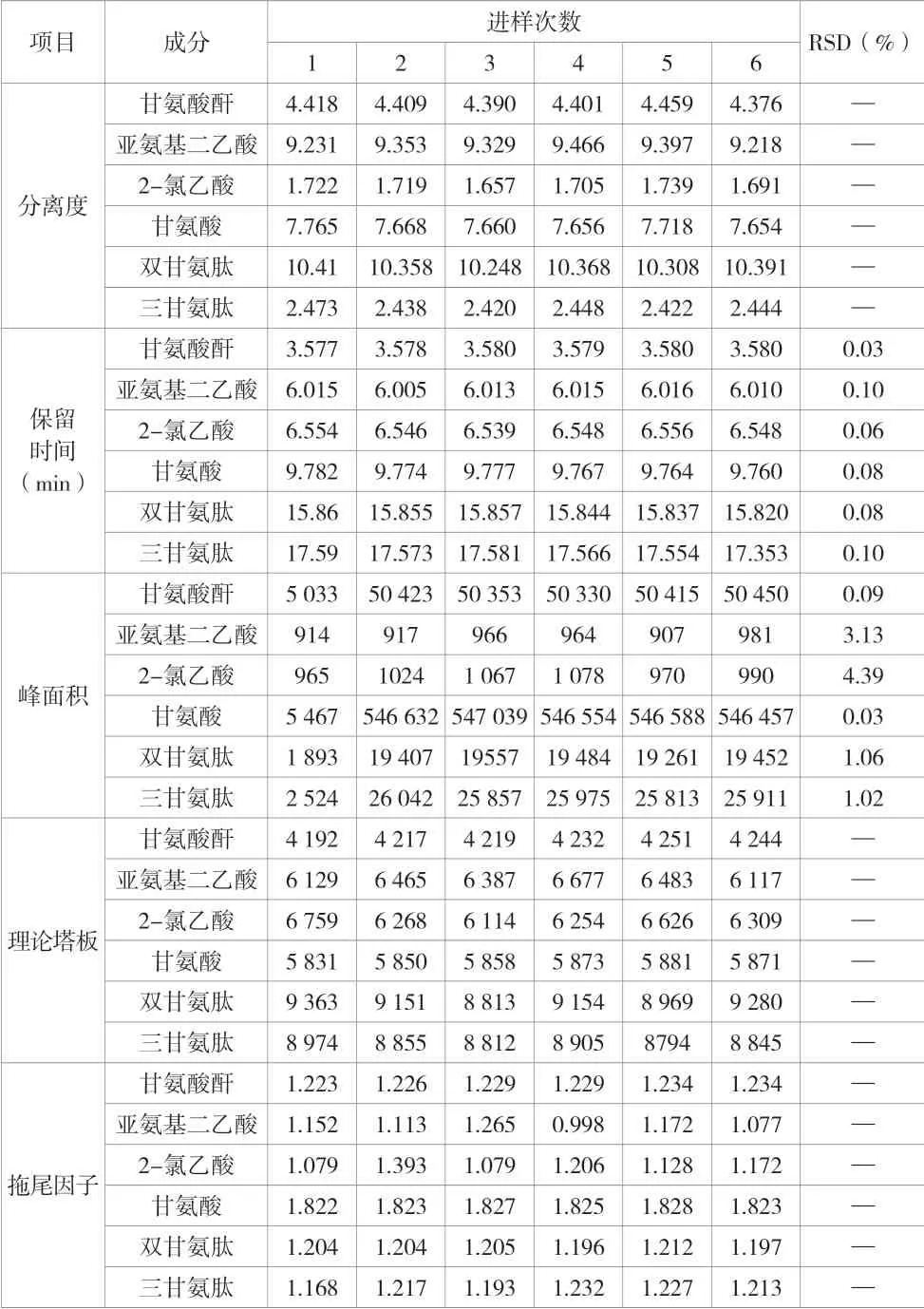
表1 系统适用性与仪器精密度试验结果
2.2 定量限
取各成分对照品溶液,用稀释剂不断稀释,精密量取20 μL 注入液相色谱仪,记录色谱图,直至峰信噪比(S/N)≥10,计算各成分定量限和定量限浓度。配制6 份定量限溶液,精密量取上述溶液各20 μL 注入液相色谱仪,记录色谱图。计算各成分峰面积RSD 和保留时间RSD。
结果表明:信噪比(S/N)≥10,6 份定量限溶液中各峰的保留时间RSD ≤1.0%,峰面积RSD ≤10%。定量限测定结果见表2。

表2 定量限测定结果
2.3 检测限
取定量限项下的定量限溶液,用稀释剂稀释,精密量取20 μL 注入液相色谱仪,记录色谱图,当信噪比(S/N)≥3,计算各成分检测限和检测限浓度。
检测限测定结果见表3。
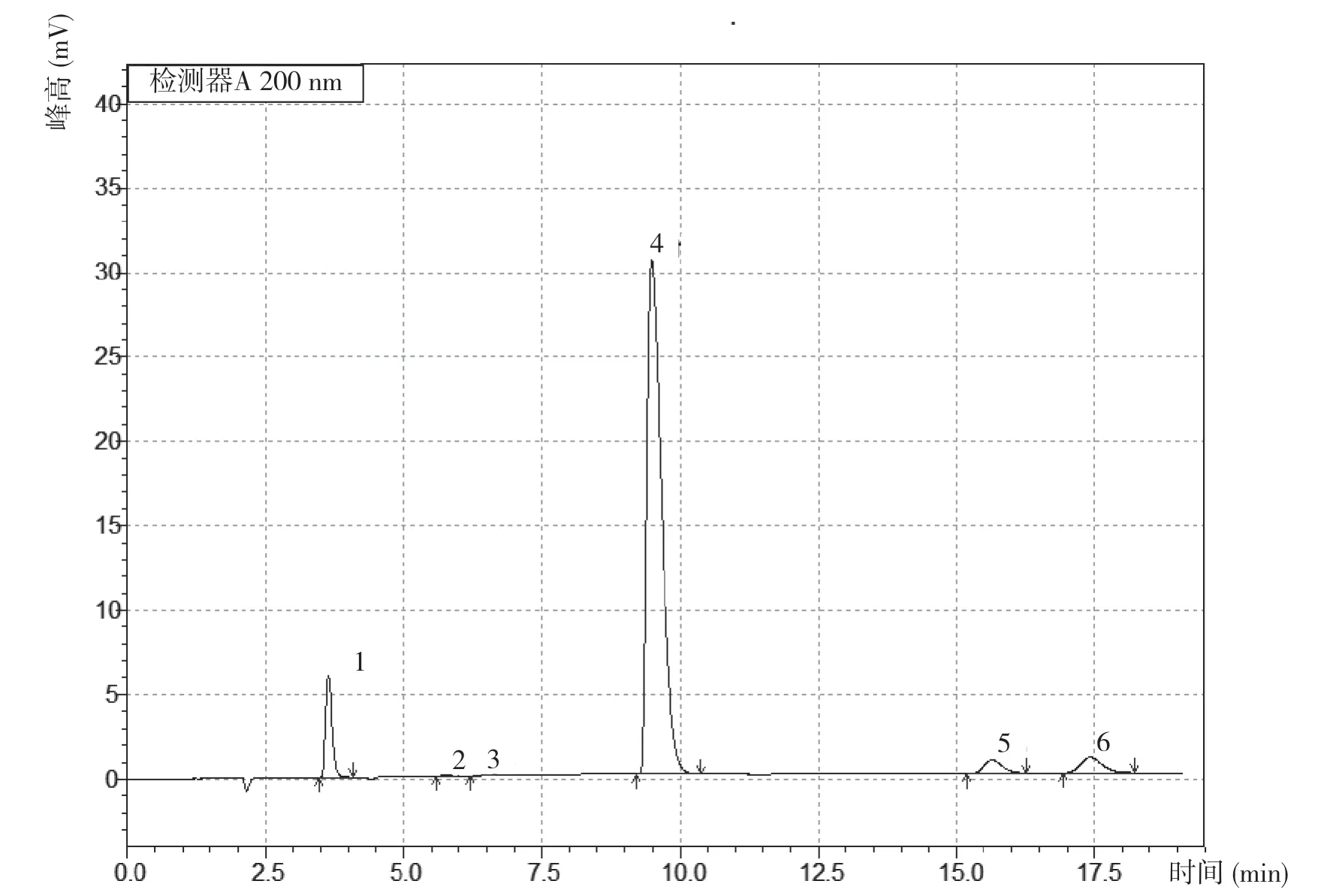
图1 液相色谱图
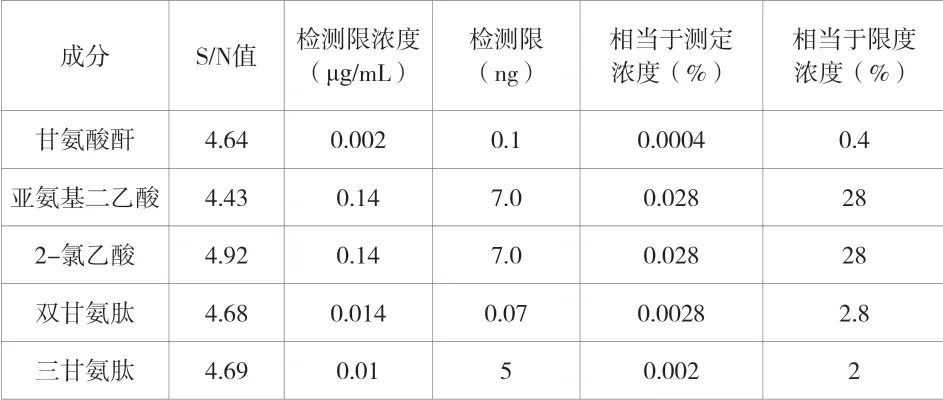
表3 检测限测定结果
2.4 线性与范围
精密量取对照品二级贮备液(25 μg/mL)0.3 mL、0.5 mL、0.8 mL、1.0 mL 和1.2 mL,分别置50 mL 量瓶中,加稀释剂稀释至刻度,摇匀。精密量取上述溶液各20 μL 注入液相色谱仪,记录色谱图。
对上述溶液的浓度(x)与相应的峰面积(y)作线性回归,计算回归方程及其相关系数R、斜率K、截距。
结果表明各组分在00.6 μg/mL~0.6 μg/mL 浓度范围内(相当于测定浓度0%~0.12%,相当于含量0%~0.12%,相当于限度浓度0%~120%),峰面积与浓度呈线性关系(R >0.998)。回归方程与线性范围见表4。
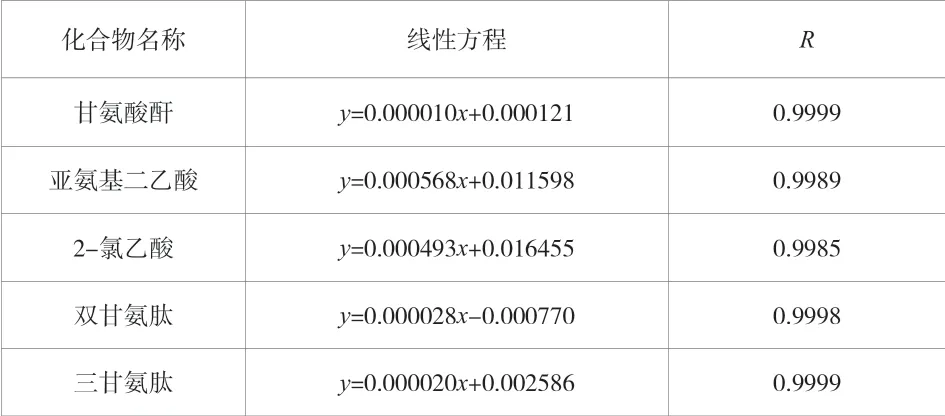
表4 各杂质回归方程及相关系数
2.5 准确度
按照“1.3”介绍的方法,分别取供试品溶液、80%限度加标供试品溶液、100%限度加标供试品溶液、120%限度加标供试品溶液,注入液相色谱仪,记录色谱图,以外标法测定并计算含量,杂质甘氨酸酐、亚氨基二乙酸、2-氯乙酸、双甘氨肽、三甘氨肽9 次测定的平均回收率分别为100.23%、98.58%、98.68%、99.69%、99.97%,RSD 值分别为1.20%、0.33%、0.86%、1.15%、0.82%。结果表明,该方法准确度均良好。
2.6 耐用性
分别调节流速为0.8 mL/min 与1.2 mL/min、柱温20 ℃与30 ℃、pH=2.1 与pH=2.3、流动相比例缓冲液∶乙腈=98.5 ∶1.5 和缓冲液:乙腈=96.5 ∶3.5 时,进样对照品溶液,结果表明,在上述各条件下进行试验,甘氨酸色谱峰与各杂质分离度均大于1.5,该方法耐用性良好[6]。
2.7 溶液稳定性
取供试品溶液于25 ℃自动进样器中,按照“1.3”介绍的方法,分别在0 h、2 h、4 h、6 h、8 h 进样分析,考察供试品溶液的稳定性。结果表明,各杂质在8 h 放置时间内,含量相对稳定。因此,供试品溶液配制完成后可在8 h 内测定。
2.8 供试品有关物质测定
取该品3 批,按照“1.2”介绍的供试品溶液的配制方法,按照“1.3”介绍的方法进行测定,杂质甘氨酸酐、亚氨基二乙酸、2-氯乙酸、双甘氨肽、三甘氨肽以外的标法计算有关物质含量,其他单个最大杂质与总杂质以面积归一化法测定,结果见表5。结果显示,3 批甘氨酸有关物质测定结果均符合要求。
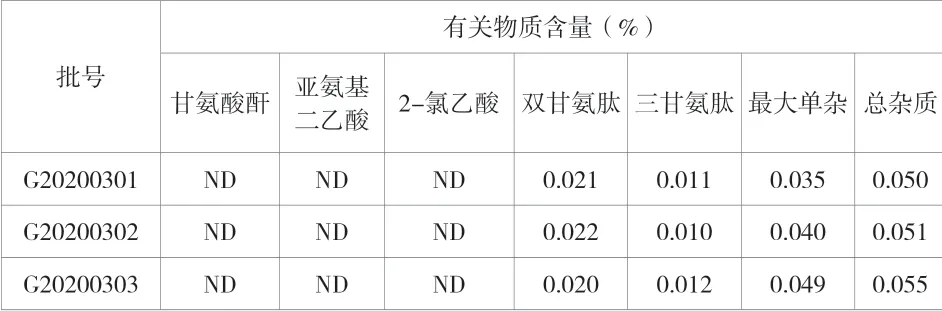
表5 甘氨酸有关物质测定结果
3 讨论
该研究对甘氨酸中有关物质的检测条件进行了优化,筛选了不同类别的色谱柱及流动相条件,最终以CenturySIL C18-AQ 作为分析柱时,色谱峰出峰快,分析周期短,基线平稳,且分离度各项指标满足要求,色谱柱廉价、节约成本。由于甘氨酸及各杂质均为两性化合物,分子极性较大,在C18 固定相中的保留较弱,在方法开发的过程中,加入了离子对试剂庚烷磺酸钠,其庚烷基与色谱柱的固定相吸附结合,暴露出磺酸基负离子,吸附流动相中的阳离子被分析物,增强阳离子化合物的保留。该试验中通过调节流动相pH 值,实现了甘氨酸和各杂质的有效分离。同时研究还发现,随着柱温的升高,各成分出峰时间缩短,柱温为20 ℃、25 ℃、30 ℃时,各色谱峰之间均能满足分离的要求,综合考虑选定柱温为25 ℃。随着流速的增加,各成分出峰时间缩短,各色谱峰之间满足分离的要求,选定流速为1.0 mL/min。该方法简便、准确,能够为甘氨酸的质量控制提供依据。
猜你喜欢
科学与财富(2021年35期)2021-05-10
理化检验-化学分册(2021年1期)2021-03-06
井冈山大学学报(自然科学版)(2021年1期)2021-03-05
核化学与放射化学(2020年4期)2020-08-21
食品安全导刊·中旬刊(2020年5期)2020-06-04
湿法冶金(2018年2期)2018-04-25
科技传播(2016年15期)2016-11-30
质谱学报(2016年4期)2016-08-02
应用化工(2014年7期)2014-08-09
中国氯碱(2014年11期)2014-02-28
