
自催化型石墨烯金纳米探针的制备及用于非小细胞肺癌CYFRA211电化学检测
2020-07-14 02:35李瑞怡储红霞余敏仪李姝晗李在均
分析化学 2020年7期
李瑞怡 储红霞 余敏仪 李姝晗 李在均


摘要早期诊断对提高肺癌患者生活质量和延长生存期至关重要。本研究通过热解柠檬酸与谷氨酰胺的混合物得到谷氨酰胺功能化石墨烯量子点(GlnGQD),所制备的GlnGQD与HAuCI4反应形成GlnGQD/Au复合物。GlnGQD作为还原剂将Au3+转化为Au0,最终形成Au纳米晶体。GlnGQD还作为稳定剂被固定于Au纳米晶体的表面,从而实现GlnGQD与Au的杂交。扫描电镜、透射电镜、X射线衍射和红外光谱分析表明,GlnGQD/Au平均粒径为(31.2±0.15) nm,具有立方金的晶体结构,Au和N元素均匀分布于球形粒子表面,含有丰富的OH、NH、COOH等功能基团。将发夹DNA 2 (H2) 通过AuS键连接到Au纳米粒子表面,再利用EDC/NHS活化实现GlnGQD的COOH与硫堇(Thi)的氨基缩合,得到H2GlnGQD/AuThi氧化还原探针,基于此构建了无酶放大电化学传感平台。在CYFRA211存在下,此传感平台的H2可与预先修饰在金电极表面的发夹DNA 1(H1)杂交,释放出一个CYFRA211分子,所释放的CYFRA211可直接用于下一次靶DNA循环。通过靶诱导的DNA自组装链式反应,一个CYFRA211靶分子可將多个氧化还原探针固定在金电极表面,从而产生显著的电化学信号放大效应。H2与H1的杂交实现对靶DNA的特异性响应,Thi在电极表面发生可逆性氧化还原反应, 对靶DNA的响应产生电化学信号,而GlnGQD/Au原位催化Thi的氧化还原反应将进一步放大检测信号。 CYFRA211浓度在2~100000 fmol/L之间,其差分脉冲伏安峰电流随CYFRA211浓度增加而线性增大。本方法检出限为0.67 fmol/L(S/N=3), 灵敏度明显优于文献报道的方法。本方法成功用于人血清中CYFRA211的电化学检测。
关键词石墨烯金复合物; 氧化还原探针; 自催化作用; 电化学传感器; 肺癌诊断
1引 言
肺癌是全球发病率和死亡率最高的恶性肿瘤[1~4]。由于生活方式和社会经济发展的多样性,肺癌发病率呈现明显的地理和性别差异[3]; 此外,肺癌发病还与吸烟、空气污染和职业等因素有关[4,5]。目前,术后肿瘤治疗的主要手段是化疗和放疗,但化疗和放疗处理对正常细胞具有严重的毒副作用[6],早期诊断仍是肺癌患者提高生活质量和延长生存期的最佳方案。细胞角蛋白19片段211(CYFRA211)是细胞角蛋白19的可溶性片段,在细胞凋亡过程中释放到血液中,是恶性肿瘤的常用重要生物标志物[7]。研究表明,肺癌患者的CYFRA211水平与癌胚抗原和神经元特异性烯醇化酶的转移和活性的关联性强,近50%患者血清CYFRA 211处于高水平表达[8]。因此,血清CYFRA211成为肺癌早期诊断的重要参考指标[9]。
近年来,多种分析技术被用于检测与癌症相关的生物标记物,如蛋白免疫印迹法[10]、免疫细胞化学法[11]、流式细胞术[12]、PCR[13]和电化学传感器[14]等。蛋白免疫印迹是分子生物学、生物化学和免疫遗传学研究常用的实验技术,通过特异性抗体对凝胶电泳处理后的细胞或生物组织样品进行染色,然后基于着色的位置和深度,分析特定蛋白质在细胞或组织中的表达情况。该方法灵敏度较低,而且需要使用昂贵仪器[15]。免疫细胞化学是用标记后的抗体或抗原对细胞相应抗原或抗体进行定性、定位或定量检测的方法。该方法过程繁琐,且分析周期长[16]。PCR是临床上识别和定量分析DNA和RNA的常规方法。然而,采用传统PCR技术检测复杂DNA混合物中的稀有变异拷贝时只能得到一种平均信号,方法灵敏度较低; 此外,PCR操作较为复杂,需要熟练的专业技术人员[17]。近年来,电化学传感器检测与癌症相关的DNA序列引起极大关注。随着纳米技术和基于DNA信号放大策略的快速发展,电化学传感技术在灵敏度、选择性和重现性方面得到进一步提高,成为临床分析和相关研究的重要手段。然而,肺癌早期的CYFRA 211浓度极低,现有电化学传感器仍不能满足早期诊断对灵敏度的需要。
纳米金和石墨烯是电分析化学领域常用的纳米材料。纳米金导电性好、比表面积大、生物相容性好,且具有独特的催化作用[18]。石墨烯是由六边形sp2碳原子形成的二维碳材料,具有比表面积大、导电性好、载流子迁移快等优点[19]。最近,金和石墨烯的杂合物成为材料领域的研究热点[20]。石墨烯金杂交物提供了较单独纳米金和石墨烯更高的催化活性[21]。石墨烯量子点不同于经典石墨烯,由尺寸仅为数纳米的石墨烯片组成,具有更大的比表面和更多的功能基团。因此,将纳米金和石墨烯量子点结合可进一步提高杂交物的功能性和催化活性[22]。
1电化学检测
基于DNA的信号放大策略主要包括酶辅助核酸扩增和无酶扩增策略。酶辅助核酸扩增策略常与含有聚合酶、核酸外切酶或内切核酸酶的蛋白质核酸酶单独或组合使用[23],然而,酶调节的信号放大可能产生一些非特异性反应,增大了假阳性发生的几率[22]。近年来,无酶扩增信号放大策略受到青睐,具有更好的稳定性、重现性和可靠性。无酶放大策略主要包括催化发夹DNA组装[24]、杂交连锁反应[25]和立足点触发链置换反应[27]。通过DNA序列的合理设计,催化发夹组装可方便地实现对不同靶DNA的特异性电化学响应,具有高效和稳定的优势,特别适合构建生物传感器 [27]。基于纳米金石墨烯量子点的特性,将其融入到催化发夹DNA组装无酶放大策略,有望实现血清CYFRA211的超灵敏电化学检测。
本研究采用热解柠檬酸与谷氨酰胺(Gln)的混合物得到了谷氨酰胺功能化石墨烯量子点(GlnGQD),与HAuCl4反应形成GlnGQD/Au杂交物。通过共价健将发夹DNA 2 (H2)和硫堇(Thi)固定到杂交物的表面,得到H2GlnGQD/AuThi自催化型氧化还原探针,用于构建无酶放大电化学传感器。由于GlnGQD/Au优异的电催化活性和靶诱导的高效无酶循环扩增反应,此电化学传感器具有超高的灵敏度、选择性和稳定性,并成功应用于血清中CYFRA211的电化学检测。
2实验部分
2.1仪器与试剂
HITACHI S4800场发射扫描电子显微镜(SEM, 日本日立高新公司); JEM2100(HR)透射电子显微镜(TEM, 日本JEOL公司); D8 Advance X射线衍射(XRD, 德国布鲁克公司); Nicolet iS50 FTIR 傅立叶红外光谱仪(美国赛默飞世尔科技公司); CHI660D电化学工作站(上海辰华公司)。采用三电极系统:Ag/AgCl电极(饱和KCl)、铂丝和裸金电极或修饰电极(直径2 mm)分别作为参比电极、对电极和工作电极。 差分脉冲伏安法(DPV)测量电位范围为0.6~0.2 V,步进电位为4 mV,频率为25 Hz,幅度为25 mV; 电化学阻抗谱(EIS)测量的电位幅度为±5 mV,频率范围0.01~105 Hz。电化学测量均在室温下进行,期间保持N2气氛。
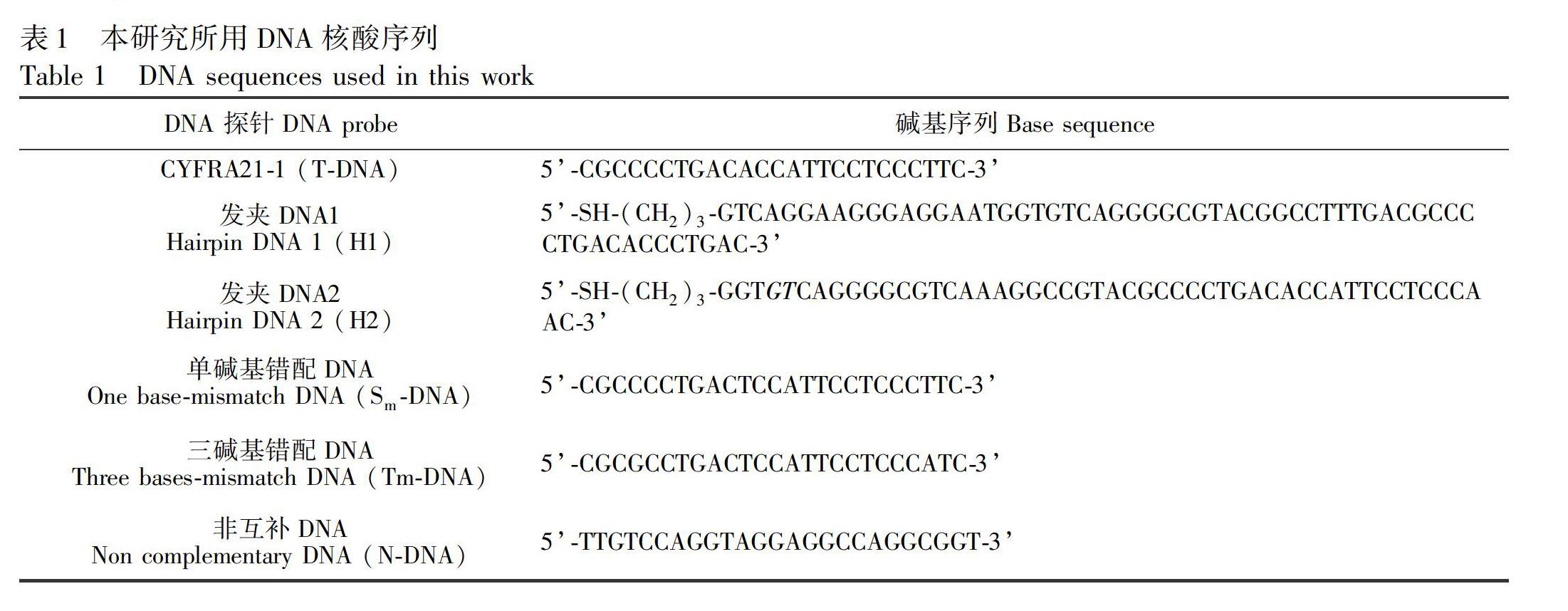
柠檬酸、Gln、氯金酸(HAuCI4)、Thi、三(2羧乙基)膦盐酸盐(TCEP)和6巯基己1醇(MCH)(Sigma公司)。0.01 mol/L磷酸盐缓冲溶液(Na2HPO4KH2PO4NaClKCl,PBS,pH 7.4)。含有20 mmol/L TrisHCI和0.001 mol/L MgCl2/0.001 mol/L CaCI2/5 0.001 mol/L KCI(pH 7.4)的Tris/Mg/K緩冲液制备DNA储备溶液,在20℃储存,备用。使用前,发夹DNA 1(H1)和DNA 2(H2)在95℃加热10 min,然后以1℃/min缓慢冷却至室温,形成稳定的发夹结构。所用DNA核酸序列由生工生物工程(上海)股份有限公司合成,碱基序列见表1。其它试剂至少为分析纯,购自上海化学公司。实验用水为MilliQ纯水系统纯化的超纯水(18.2 MΩ cm)。
2.2GlnGQD/Au合成
按1∶0.5摩尔比的柠檬酸和谷氨酰胺混合物,加适量水充分溶解,在80℃下蒸干,然后转移至高温烘箱中,180℃热解3 h,得到谷氨酰胺功能化石墨烯量子点(GlnGQD)。
将氯金酸溶液(0.05 g/L, 49 mL)加热至沸腾,然后加入GluGQD溶液(50 g/L, 1.0 mL),保持煮沸条件下反应至溶液变为酒红色,迅速冷却至室温,8000 r/min离心8 min,得到GlnGQD/Au杂交物粗品。收集的固体用水洗涤3次,重新分散在1.0 mL TrisHCl缓冲溶液(pH 7.4, 0.05 mol/L)中,得到GlnGQD/Au储备溶液。
退火处理后的H2溶液(100 μmol/L,20 μL)与TrisHCl缓冲液(20 mmol/L,50 μL,pH 7.4)混合。加入TCEP溶液(2 mmol/L,40 μL)后,搅拌1 h,通过还原断开因H2暴露在空气中氧化产生的二硫键,实现H1探针中巯基的活化[28]。在搅拌下加入GlnGQD/Au储备溶液(0.6 mL)。将2 mol/L NaCl和1% SDS分别加入到上述混合溶液中,使最终溶液中SDS和NaCl的浓度分别为0.01%(w/V)和0.16 mol/L。 将此混合溶液常温孵育24 h, 16000 r/min离心10 min。收集到的沉淀用TrisHCl缓冲液(pH 7.4)洗涤后,重新分散在pH 7.4的TrisHCl缓冲液中。
将H2GlnGQD/Au超声分散到1.0 mL TE缓冲溶液,加入EDC溶液(20 mg/mL, 10 μL)和NHS溶液(20 mg/mL, 5 μL), 37℃振荡(250 r/min)孵育90 min。将活化后的H2GlnGQD/Au与Thi溶液(0.1 mmol/L)混合, 25℃振荡孵育(250 r/min)过夜, 16000 r/min离心10 min。收集到的H2GlnGQDThi用TrisHCl缓冲液(pH 7.4)洗涤3次,重新分散在TrisHCl缓冲液中, 得到H2GlnGQDThi储备液, 4℃储存,备用。
2.4修饰电极的制备
等体积的H1溶液(10 μmol/L)和TCEP溶液(1mmol/L)混合,搅拌1 h,通过TCEP还原断开二硫键,实现H1分子中巯基的活化,用TrisHCl缓冲液稀释得到0.3 μmol/L H1储备溶液。将5 μL H1储备溶液滴加到预处理的金电极表面, 在37℃孵育2 h后,修饰电极用10 mmol/L PBS(pH 7.4,0.1 mol/L NaCl)洗涤,氮气吹干,然后滴加5 μL MCH溶液(1 mmol/L),室温孵育1 h,用pH 7.4的PBS溶液洗涤,氮气吹干,于4℃储存,备用。
2.5电化学检测CYFRA211
将CYFRA211标准溶液或血清样品与H2GlnGQDThi储备溶液混合,室温下孵育20 min。将5 μL上述混合溶液滴到已制备的修饰电极表面,室温下孵育80 min,用10 mmol/L PBS(pH 7.4)洗涤3次,在电化学工作站上测定DPV曲线,电解质溶液为10 mmol/L PBS(pH 7.4,含1.0 mol/L NaCl)。
3结果与讨论
3.1H2GlnGQDThi氧化还原探针的合成与表征
H2GlnGQDThi氧化还原探针的合成方案见图1。首先,通过柠檬酸分子间脱水缩合及随后的碳化形成由六边形sp2碳原子组成的二维石墨烯片,再利用Gln分子中的氨基与石墨烯片边缘的羧基脱水实现GQD的功能化[29]。采取干法热解制备GlnGQD,石墨烯片的形成与功能化通过一步热解反应即可完成,方法简单、高效、环保。然后,GlnGQD与HAuCl4反应合成GluGQD/Au。一方面,GlnGQD作为还原剂,将Au3+转化为Au0,最终形成Au纳米晶体; 另一方面,GlnGQD作为稳定剂,通过其功能基团与Au纳米晶体表面的Au+配位形成稳定的亲水包覆层[30, 31]。Au纳米晶体是良好的导体,而GlnGQD为典型的半导体,因此,二者结合将在界面上产生类似金属/半导体二极管的结构。通过二极管对电化学检测信号的放大作用,可提高分析方法的灵敏度。
测定了反应体系的紫外可见吸收光谱和荧光光谱。如图2A所示,GlnGQD溶液在可见光区域的吸收很弱,无特征吸收峰。加入HAuCl4后,体系在540 nm处出现一个明显的吸收峰,且峰吸光度值随反应时间的延长而迅速增大。由于反应体系中只有纳米Au可产生特征的可见光区,加入HAuCl4后,体系出现540 nm处吸收峰,表明体系中形成了Au纳米晶体,同时也表明GlnGQD能还原Au3+为Au0,最终导致形成纳米Au。随着反应时间延长,形成的Au纳米晶体数量迅速增大,导致可见区的吸光度增大。当反应时间超过1 min,吸光度值趋于稳定,说明生成Au纳米晶体的反应接近完全。由图2B可见,加入HAuCl4后,GlnGQD的荧光强度明显下降。这是因为Au纳米晶体有良好的导电性,与GlnGQD结合,将产生一个快速的能量转移过程,导致GlnGQD 荧光猝灭。随着反应时间延长,所形成的金纳米晶体数量增多,荧光猝灭程度增大。当反应时间超过1 min,体系荧光强度变得十分微弱,并趋于
通过AuS键将发夹DNA 2 (H2) 固定在GlnGQD/Au纳米粒子表面。考察了GlnGQD/Au在不同浓度H2溶液中孵育24 h的吸收光谱。由图4可见,GlnGQD/Au在350nm处有一个较强的吸收峰,是GlnGQD/Au中GlnGQD的吸收。当它在H2溶液中孵育后,260 nm处的吸光度(A260 nm)明显增加。由于DNA的特征紫外吸收峰位于260 nm,上述结果也证明DNA已成功连接到了Au纳米晶体表面。随着H2浓度增加,A260 nm值迅速增大,表明更多的H2固定到了Au纳米晶体表面。H2浓度大于100 μmol/L时,A260 nm值趋于稳定,表明Au与H2之间的反应达到平衡。本研究选择100 μmol/L H2制备氧化还原探针。采用EDC/NHS活化GlnGQD的COOH,与Thi的氨基缩合,得到H2GlnGQD/AuThi氧化还原探针。
3.2CYFRA211电化学传感平台
H2GlnGQDThi作为一个自催化型氧化还原探针,用于构建CYFRA211电化学传感平台(图5)。首先,将发夹DNA1(H1)通过AuS键固定于金电极表面,用MCH对金电极表面剩余活性位点进行封闭处理。在靶DNA(CYFRA211)存在下,靶DNA与电极表面的H1通过碱基配对进行杂交,导致H1的发夹结构部分打开,能与H2配对的碱基序列暴露。通过碱基链置换反应,H2GlnGQDThi与H1杂交产生一个游离靶DNA。释放的靶DNA继续打开电极表面处于发夹状态的H1,打开的H1与H2GlnGQDThi杂交, 重新释放出靶DNA。通过这种靶DNA诱导的催化发夹DNA自组装过程,一个靶DNA可以导致很多个H2GlnGQDThi探针结合到电极表面,显著放大检测信号。在H2GlnGQDThi中,H2可特异性地与被靶DNA打开发夹结构的H1结合,因此传感平台能对CYFRA211产生特异性识别。Thi能在电极表面发生可逆性氧化还原反应,对传感平台的识别产生可检测的电化学信号。GlnGQD/Au具有优异的催化活性,可原位催化Thi的氧化還原反应,实现对检测信号的放大。基于以上设计,所构建的传感平台可对CYFRA211进行高灵敏和高特异性的电化学检测。
为检验本方法测定CYFRA211的可行性,考察了构建过程中电化学传感平台的循环伏安(CV)和EIS行为。由图6可见,裸金电极具有较高的CV电流和较小的电荷转移电阻(Rct=117.3Ω)。裸金电极表面存在大量金原子,对K3Fe(CN)6的氧化和还原反应具有显著的催化作用,导致电极反应加快,从而导致CV电流的增加和Rct的下降。相对于裸金电极,H1修饰电极的CV电流明显减小,Rct增加(371.8 Ω)。这是由于H1带负电荷的磷酸骨架对Fe(CN)46产生静电排斥而难以接近电极表面,另一方面,H1的存在堵塞了部分电极与电解质之间进行电子/离子传输的通道。当用MCH封闭电极后,电极表面更多的活性位点被MCH所占据, Ret(1169 Ω)大幅增加。 不存在靶DNA时,H2GlnGQDThi不能与电极上的H1结合进入电极的表面,传感器的CV响应和Ret值(1122 Ω)基本保持不变。靶DNA存在时,由于H2GlnGQDThi被引入到电极表面,CV电流明显增加,Rct值大幅度下降(468.4 Ω), 由于H2GlnGQDThi作为自催化型氧化还原探针产生了灵敏的电化学响应。上述结果还表明,只有靶DNA才能开启发夹自组装链式反应,实现对CYFRA211的高灵敏和高特异性电化学响应。
3.3检测条件的优化
考察了H1浓度和孵化时间对DPV峰电流的影响。如图7所示,当H1浓度小于0.5 μmol/L时,DPV峰电流值随H1的浓度增加而迅速增大。当H1浓度低时,固定在电极表面的H2GlnGQDThi探针数量主要取决于电极表面H1的数量。随着H1浓度增加,固定在电极表面的H2GlnGQDThi探针数量将增多,从而产生更大的DPV电流。当H1浓度达到0.5 μmol/L时,DPV响应趋于稳定。因为H1的数量与通过靶诱导的DNA循环反应在60 min内能固定到电极表面的H2GlnGQDThi数量接近。继续增加H1浓度也不能继续增加固定在电极表面的H2GlnGQDThi数量。由图7B可见,当孵化时间小于60 min时,DPV峰电流随孵化时间的延长而迅速增大。当孵育时间超过60 min,峰电流不再增加, 说明电极表面H1已完全与H2GlnGQDThi杂交。后续实验选择0.5 μmol/L H1孵育60 min。
3.4方法的分析性能
電化学传感平台在不同浓度CYFRA211下的DPV行为见图8。随着CYFRA211浓度的增加,修饰电极的DPV电流逐渐增大(图8A)。DPV峰电流与CYFRA211浓度对数值的关系曲线如图8B所示。当靶DNA浓度在2~100000 fmol/L之间,DPV峰电流(Ip)与CYFRA211浓度的对数值(lgCDNA)呈现良好的线性关系,线性回归方程为Ip= 314.43lgCDNA +1233.3(R2=0.9988),方法检出限(S/N=3)为0.67 fmol/L。所构建的电化学传感器测定CYFRA211具有高的灵敏度,优于现有文献报道的同类传感器[32~36]。
采用相同的方法制备10个传感器,测定1.0×1012mol/L CYFRA211响应电流的相对标准偏差(RSD)为3.3%,表明传感器制备重现性良好。 采用同一个传感器测定1000 fmol/L CYFRA211,20次重复测量的RSD为2.8%,表明传感器重现性好。将制备的传感器在4℃储存30 d后,传感器对1.0×1012mol/L CYFRA211的DPV峰电流响应为原始响应值的974%以上,表明此传感器具有高的长期稳定性。
选择单碱基错配DNA序列(SmDNA)、三碱基错配DNA序列(TmDNA)和非互补DNA序列(NDNA),测试传感器对靶DNA(TDNA)CYFRA211的特异性。由图 9可见,NDNA引起的DPV峰电流值与空白PBS相当,说明传感平台对NDNA不能识别。相对于NDNA,TmDNA和SmDNA引起了明显的DPV峰电流响应。TmDNA和SmDNA与TDNA仅存在少量碱基序列差异,同样具备开启催化发夹DNA自组装过程的功能,引起较小的电化学响应。然而,碱基错配将大幅度降低TmDNA或SmDNA与H1杂交反应速率,严重制约靶诱导催化发夹DNA自组装过程的开启,从而导致引入到电极表面的H2GlnGQDThi探针数量很少,因此仅引起较小的DPV电流响应。由图9可见,TDNA产生的DPV峰电流值明显高于其它DNA序列,表明此电化学传感器测定CYFRA211具有良好的选择性。
3.5血清样品CYFRA211测定
将构建的电化学传感平台用于人血清样品中CYFRA211的测定,血清样品中未检出CYFRA211,200 fmol/L水平的加标回收率为100.4%,表明本方法具有较高的准确性。
4结 论
针对肺癌早期诊断技术灵敏度不足的问题,本研究设计合成了含发夹DNA 2、GlnGQD/Au和Thi的自催化型氧化还原探针,结合无酶放大策略,构建了CYFRA211电化学传感平台。探针的发夹DNA 2(H2)对靶DNA(TDNA)的识别产生特异性响应,利用Thi的氧化还原反应对识别靶DNA的响应产生电化学信号,GlnGQD/Au原位催化Thi的氧化还原,实现响应信号的放大。自催化氧化还原探针的设计,可改善电化传感器的特异性和灵敏度; 此外,自催化型氧化还原探针与无酶放大策略的结合进一步提高了方法的灵敏度。基于氧化还原探针和无酶循环扩增的双重信号放大,所构建的电化学传感平台对CYFRA211灵敏度高,有望在肺癌早期诊断中得到应用。
References
1Cao M M, Chen W Q. Thoracic Cancer, 2019, 10(1): 3-7
2Mizuguchi S, Nishiyama N, Iwata T, Nishida T, Izumi N, Tsukioka T, Inoue K, Kameyama M, Suehiro S. Ann. Thorac. Surg., 2007, 83(1): 216-222
3Han X, Gao Y H, Ma J M, Sang M J, Zhou S, Huang T, Mao X X. Environ. Sci. Pollut. Res., 2019, 26(10): 10083-10096
4Wang L, Liu N, Ma Z. J. Mater. Chem. B, 2015, 3(14): 2867-2872
5Chen W, Zheng R, Zeng H, Zhang S. Thorac. Cancer, 2015, 6(2): 209-215
6Mazzone P, Mekhail T. Respir. Med., 2012, 106(4): 473-492
7Boeck S, Wittwer C, Heinemann V, Haas M, Kern C, Stieber P, Nagel D, Holdenrieder S B. J. Cancer, 2013, 108(8): 1684-1694
8Kanaji N, Bandoh S, Fujita J, Ishii T, Ishida T, Kubo A. Lung Cancer, 2007, 55(3): 295-302
9Xu Y T, Xu L, Qiu M, Wang J, Zhou Q, Xu L, Wang J, Yin R. Sci. Rep., 2015, 5: 9444
10WalentowiczSadlecka M, Dziobek K, Grabiec M, Sadlecki P, Walentowicz P, Mak P, Szymankiewicz M, Kwinta P, DutschWicherek M. Am. J. Reprod. Immunol., 2019, 81: e13070
