
主客体作用在生化分析中的应用研究进展
2020-07-14 02:35崔琳赵敏惠张春阳
分析化学 2020年7期
关键词:评述
崔琳 赵敏惠 张春阳


摘要主客体作用是主体和客体在满足结构互补和能量匹配的条件下,通过非共价相互作用选择性结合,形成具有特定功能的超分子的过程。生物体系中的抗原抗体、DNA蛋白质、酶底物等生物分子之间的识别都建立在非共价作用识别的基础上。主客体识别反应具有反应条件温和、反应过程动态可逆等特点,不仅可有效克服共价结合的局限性,而且能在分子水平上模拟生物功能。本文综述了主客体作用在生化分析中的应用的最新研究进展,并对基于主客体作用的传感器的发展趋势做了展望。
关键词主客体作用; 生物传感器; 超灵敏检测; 生化分析; 评述
1引 言
主客体作用是主体和客体在满足结构互补和能量匹配等条件下,通过非共价相互作用选择性结合形成具有某种特定功能的超分子的过程[1]。非共价相互作用包括范德华力、静电引力、疏水作用和氢键等,是产生主客体识别作用的关键[2]。生物体系中抗原抗体、DNA蛋白质、酶底物等生物分子之间的识别都建立在非共价作用的识别基础上,在生物分子固定、生物传感及生物成像等领域得到广泛应用[3~5]。主客体作用在生化分析中的应用主要是以电化学和荧光分析为手段,通过主体(如冠醚、环糊精和杯芳烃)与目标物(客体)的特异性结合,实现对生物分子的定性与定量分析[6~10]。主客体作用具有高度选择性与动态可逆性等特点,广泛应用于生物活性分子的快速超灵敏检测[11~13]。近年来,研究者相继成功构建了多种基于主客体体系的传感器,展现了广阔应用前景[11~15]。本文系统总结了主客体识别的策略以及主客体作用在蛋白质、核酸、生物小分子、金属离子和异构体检测中的应用,并对主客体作用在生化分析中的发展前景进行了展望。
2主客体识别策略
基于主客体识别策略的传感器可选择性地与目标物结合,受体(主体)和目标物(客体)之间的相互作用产生的变化可通过传感器转换成可量化的信号。基于主客体作用的传感器主要包括电化学(电势、安培和电导)和光学(吸光度、荧光和化学发光)传感器[16~18],受体包括生物活性高分子(例如酶和抗体(生物传感器))和超分子主体(例如环糊精、冠醚和杯芳烃等(化学传感器))[19]。
2.1基于目标分子直接响应的生物传感器
主体和客体可直接通过非共价相互作用产生信号变化。当主体或客体含有氧化还原活性或光活性等功能基团时,主客体作用的发生就会改变相应的信号(如氧化还原电位或吸收光波长),通过这些功能基团的信号变化可准确判断是否发生了主客体作用和具体的客体分子。近年来,以冠醚为主体的荧光传感器已相继开发出来[15]。荧光传感器可通过将荧光基团结合到氮杂冠醚上而构建,氮原子上的孤对电子易向激发态荧光活性物质转移。在光致电子转移过程中,当无目标金属阳离子存在时,传感器处于闭合状态,无荧光产生。当引入金属离子后,金属离子与冠醚形成主客体络合物。氮杂冠醚的氮原子参与配位,孤对电子被束缚,阻碍了电子转移,系统处于打开状态,导致荧光恢复[20]。偶氮苯分子在有无光照的状态下可发生顺反结构互变,对环糊精的选择性结合具有明显差异。
电化学传感器通常需要电活性分子(例如亚甲基蓝(MB)、二茂铁(Fc)和硫堇(Thi)等)参与产生电信号[21]。主客体识别过程中,伴随着电子从施加的电位转移,电化学信号(例如电压、电流和阻抗等)发生变化。线性扫描伏安法是一种常见的电化学测量方法,用于测量氧化还原电活性分子在电位变化下被氧化或还原的电流变化,其电流峰波的形状和出峰位置可用于分析客体分子的作用机制[21]。
2.2基于竞争分子响应的生物传感器
由于主客体结合过程的可逆性以及不同客体分子对同一主体分子结合常数的不同,结合力强的客体分子可竞争结合力弱的客体分子与主体分子结合。Zhu等[22]利用罗丹明B和1氨基吡对环糊精(βCyclodextrin, βCD)的主客体作用差异检测有机污染物1氨基吡。罗丹明B作为探针可进入环糊精的空腔,然而具有较强亲和力的1氨基吡可竞争罗丹明B与βCD结合,根据罗丹明B和1氨基吡的比例电化学信号变化可检测1氨基吡。该比例电化学传感器的灵敏度比单信号电化学传感器高,在电活性有机污染物的检测方面极具应用前景。
3主客体作用在生化分析中的应用
3.1蛋白质的检测
基于抗原抗体相互作用以及酶与底物相互结合的亲和性,主客体作用可应用于检测临床样品中的蛋白质,例如前列腺特异性抗原(Prostate specific antigen,PSA) [23]、蛋白酶[24~30]、降钙素原[31],朊病毒蛋白[32]、禽白血病病毒[33]、胰岛素[34]以及肿瘤标志物(Carcinoembryonic antigen,CEA) [35,36]等。
碱基切除修复(Baseexcision repair,BER)是众多DNA修复机制之一,其中DNA糖基化酶(如尿嘧啶DNA糖基化酶(UracilDNA glycosylase,UDG))在维持基因组完整性方面起着关键作用。Zhao等[24]发展了一种基于铁嵌入的富氮碳纳米管(Iron embedded nitrogenrich carbon nanotubes,FeCN)过氧化物模拟酶和主客体识别的电化学生物传感器,可一步检测尿嘧啶DNA糖基化酶(UDG)(图1)。亚甲蓝(MB)标记的发夹DNA(Hairpin)连接到金纳米颗粒(AuNPs)可形成MBhairpin@AuNPs探针。由于AuNPs的空间效应和发夹DNA的茎环结构,MB不能进入电极上的β环糊精(βCD)空腔。当目标UDG存在时,它可从发夹DNA探针茎上去除尿嘧啶,打开发卡探针。MB与βCD之间的主客体识别使MBhairpin@AuNPs探针组装在电极表面。电解质中L半胱氨酸(LCysteine,RSH)被氧气氧化生成H2O2,FeCN模拟酶催化MB氧化,显著增强电化学信号。该传感器可超灵敏和一步測定碱基切除修复酶(UDG)活性,检出限为7.4×10U/mL。
[31]设计了一种基于主客体纳米网催化扩增检测降钙素原(Procalcitonin,PCT)的无酶电化学免疫传感器。他们用聚酰胺胺型树枝状分子(Poly(amidoamine)dendrimer,PAMAM)包封金纳米粒子作为纳米载体结合环糊精βCD,得到βCD功能化的PAMAMAu(Poly(amidoamine)dendrimerencapsulated Au nanoparticles); 同时利用二茂铁衍生物FcFc作为桥梁结合两个环糊精分子,进入βCD/PAMAMAu的疏水内腔,形成FcFc/βCD/PAMAMAu探针。二抗(Ab2)通过化学吸附作用连接到网状的纳米结构上。由于抗坏血酸具有很强的还原性,很容易被氧化成脱氢抗坏血酸,利用FcFc和PAMAMAu协同催化抗坏血酸(Ascorbic acid,AA)氧化进行无酶信号放大,同时利用电极上的抗体(Ab1)形成抗体抗原抗体三明治夹心结构,可实现降钙素原的超灵敏检测。该传感器具有灵敏度高和重现性好的优点,检测范围为1.80 pg/mL~500 ng/mL,检出限为0.36 pg/mL。
Zhao等[25]将主客体识别与三重信号放大相结合,构建了电致化学发光(Electrochemiluminescence,ECL)生物传感器,用于超灵敏检测尿嘧啶DNA糖基化酶(UDG)(图2)。一个双标记的发夹DNA探针的一端标记Fc作为客体分子,另一端标记FeMOF/AuNPs@luminol作为ECL标签。当UDG存在时,它可从FeMOF/AuNPs@luminol修饰的发夹DNA探针的茎中去除尿嘧啶,打开发夹结构,并释放鲁米诺修饰的单链DNA(ssDNA),通过Fc和βCD之间的主客体识别进一步将FeMOF/AuNPs@luminol修饰的ssDNAs组装在电极表面。FeMOF中的Fe3+可与亚铁氰化钾反应形成普鲁士蓝,催化H2O2分解,生成羟基自由基,并能进一步与鲁米诺自由基反应产生放大的ECL信号。该ECL传感器具有选择性好和灵敏度高的特点,可用于筛选UDG抑制剂和细胞中UDG活性的检测,检出限为2.5 × 10
3.2核糖核酸检测
核糖核酸的检测在癌症和病原菌检测以及法医学鉴定中起着重要作用。宿主客体之间相互作用依赖于非共价键,随着环境条件的变化,这种非共价键容易被分解,而主客体作用由于非共价性质所具有的可逆性质可实现传感器的循环利用,提高重现性。利用主客体作用可实现对核糖核酸的快速超灵敏检测[37~44]。
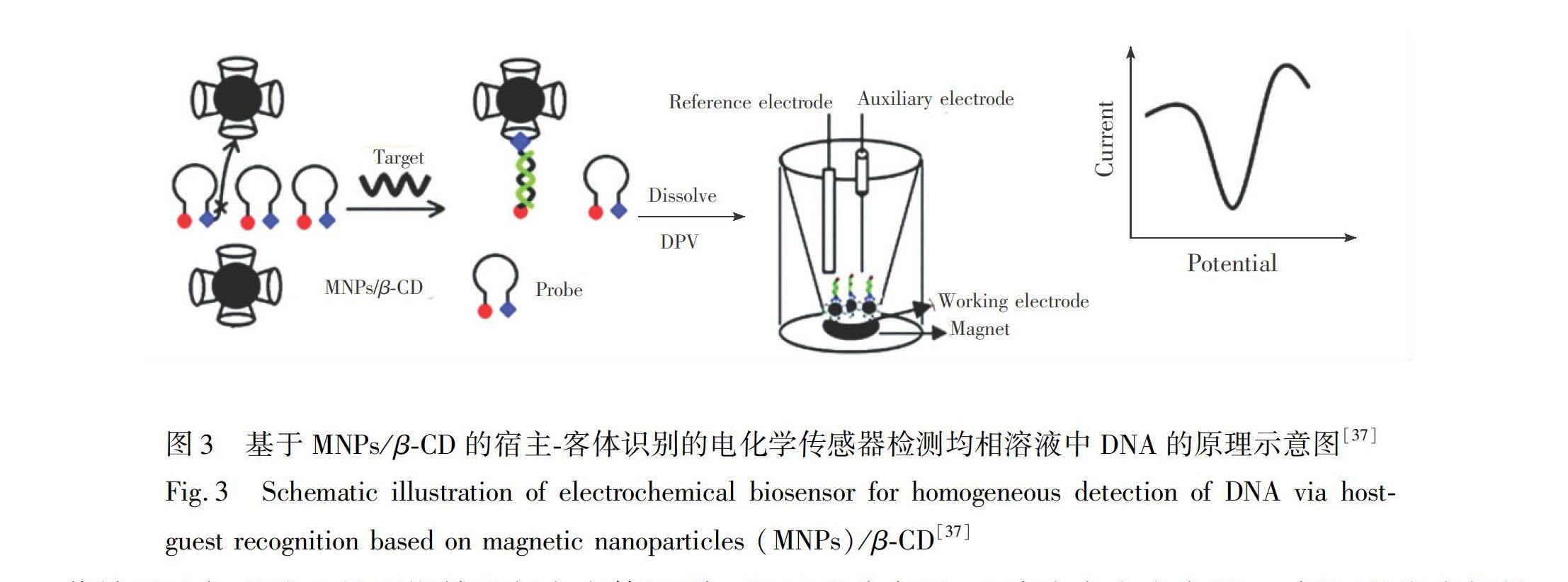
Zheng等[37]设计了一种利用磁性纳米粒子(Magnetic nanoparticles,MNPs)/环糊精进行主客体识别、分离和检测均相溶液中乙肝病毒(Hepatitis B virus,HBV)DNA的电化学传感器(图3)。他们设计了一个具有茎环结构的DNA探针用于电化学检测。该茎环结构通过碱基配对形成,该环中序列与靶HBV序列特异性互补,并在5端标记dabcyl作为客体分子,在3端标记金纳米粒子作为电化学标簽。在电极上修饰合成的βCD能固定磁性纳米颗粒(MNPs/βCD),利用主客体识别可对DNA进行检测。当无目标DNA存在时,探针处于茎环结构状态,客体分子不能被捕获和检测。当目标DNA存在时,探针展开并形成双链DNA,导致客体分子不再与金纳米粒子紧密相连,其可进一步通过环糊精与dabcyl的主客体识别被MNPs/βCD捕获,产生电化学信号。该传感器检出限为9.930×10
Yang等[38]采用三硫代碳酸盐修饰柱芳烃(Trithiocarbonate modified pillar[5]arene,P5ACTA),利用柱芳烃的主客体识别和DNA杂交技术构建了一个可回收利用的电化学传感器,用于检测乳腺癌易感基因(Breast cancer,BRCA)。他们将BRCA靶DNA(TDNA)与亚甲基蓝标记的DNA信号探针和烷基胺修饰的捕获DNA均质杂交形成三明治型DNA,同时将三硫代碳酸盐修饰柱芳烃(P5ACTA)固定在金电极上,通过P5ACTA的主客体识别捕获三明治型DNA。移入辣根过氧化物酶(Horseradish peroxidase, HRP)和H2O2显著提高了该电化学传感器的灵敏度。该电化学传感器线性范围为3.3×10mol/L,检出限为1 nmol/L。由于P5ACTA与烷基胺修饰DNA之间存在可逆的主客体作用, 该电化学传感器可通过简单的洗涤处理可回收利用。
Jiang等[39]将靶标分子触发的链转移反应(Toeholdtriggered strand displacement reaction,TSDR)诱导的目标物循坏利用和Fe3O4@SiO2@βCD的主客体作用相结合,构建了无酶均相检测DNA的电化学传感器。当无目标DNA分子时,两端均标记二茂铁(Fc)的发卡探针(Hairpin DNA,H1)茎环关闭,二茂铁无法与磁珠上的环糊精进行主客体识别。经过磁分离后,上清液中含有大量Fc标记的发卡探针H1能在电极上产生强电流信号。加入目标DNA后,触发辅助探针(Assistance probes,A1和A2)与H1杂交发生逐步分支迁移,产生更稳定的双链复合物,释放目标DNA,可实现信号的循环放大。基于目标链的循环利用可连续打开大量H1探针,通过DNA和Fe3O4@SiO2@βCD之间的主客体识别,大量Fc标记的H1探针被磁铁吸引,上清液中残留的探针减少,导致峰电流下降。该电化学传感器的线性范围宽(1~5000 pmol/L),检出限为0.3 pmol/L,具有良好的选择性。
Jiang等[40]基于氮掺杂氧化石墨烯/环糊精聚合物与Fc标记的发卡探针的主客体识别和Mg2+辅助循环裂解反应,构建了可检测目标DNA的电化学传感器(图4)。他们将热退火氧化石墨烯和三聚氰胺合成的氮掺杂氧化石墨烯(Nitrogendoped reduced graphene oxide,NRGO)分散在环糊精聚合物(βCyclodextrin polymer,βCDP)溶液中形成NRGO/βCDP纳米复合材料,用于修饰玻碳电极,构建电化学传感器。在无目标DNA存在时,电极上的βCDP不能识别发卡探针(H1),产生较小的电流信号。当目标DNA存在时,Subunit DNA S1的发卡结构打开,与发卡探针 H1组装产生Mg2 +依赖性DNA酶,其在Mg2+的作用下将Fc双标记的H1发卡探针裂解成两个单链寡核苷酸,进一步通过βCDP与Fc的主客体识别,产生增强的峰值电流。该电化学传感器具有较高的灵敏度和选择性,可用于DNA和miRNA检测,线性范围分别为0.01~1000 pmol/L和0.05~ 500 pmol/L,检出限分别为3.2 fmol/L和18 fmol/L。
3.3生物小分子的检测
许多生物小分子(如多巴胺(Dopamine)、腺苷、血清素和胆固醇等)在人体生命代谢活动中发挥了重要作用,发展超灵敏的分析方法对这些生物小分子[45~53]进行选择性检测具有重要的临床应用价值。目前利用主客体作用可实现对多巴胺[45,52]、氯霉素[46]、腺苷[47,53]、甾族化合物(胆固醇)[48]、乌头碱[49]、对硝基氯苯[50]、除草剂2甲4氯(2Methyl4chlorophenoxyacetic acid,MCPA)[51]和血清素[52]等小分子的选择性检测。
多巴胺是一种重要的神经递质,广泛分布于哺乳动物中枢神经系统,多巴胺缺失与帕金森疾病密切相关。Liu等[45]制备了三维氮掺杂石墨烯(3D nitrogendoped graphene,3DNG),利用环糊精的主客体作用,把3DNG作为βCD的电极基板建立一种新型的生物传感器,应用于多巴胺和对乙酰氨基酚(Acetaminophen, APAP)的选择性检测。三维氮掺杂石墨烯的结构类似于章鱼的触须,在它的触角上固定环糊精分子作为吸盘去捕获客体分子。该电化学传感器对多巴胺和对乙酰氨基酚的检测灵敏度分别为5468.6 μA/(mmol/L cm2) 和2419.2 μA/(mmol/L cm2)。
Wang等[46]以AuPd双金属纳米粒子(AuPd bimetallic nanoprobe)为信号标记,构建了基于环糊精和金刚烷(Adamantine,ADA)主客体作用的电化学免疫传感器,用于检测氯霉素(Chloramphenicol,CAP)(图5)。该传感器利用AuPd納米粒子对NaBH4氧化的电催化作用产生电化学信号,用于免疫分析。通过竞争免疫反应,ADA标记的抗体与固定在多壁碳纳米管修饰的电极表面的抗原CAP特异性结合,通过环糊精和金刚烷的主客体识别,将环糊精修饰的AuPd双金属纳米粒子固定在免疫传感器上,导致AuPd纳米粒子大量负载。AuPd纳米粒子能高效电催化NaBH4的氧化反应,产生电化学信号,可对氯霉素进行超灵敏检测。该传感器具有良好选择性,检测线性范围为50 pg/mL~50 μg/mL,检出限为4.6 pg/mL。
Yang等[47]基于三磷酸腺苷(Adenosine triphosphate,ATP)与羧酸二茂铁(Ferrocenecarboxylic acid,FcA)和6氨基β环糊精(Per6ammoniumβcyclodextrin,pABCD)的主客体竞争反应,利用羧酸二茂铁(FcA)作为电化学探针选择性检测三磷酸腺苷。在pABCD分子中,环糊精6号位的主羟基被氨基取代,由于pABCD的空腔与腺苷碱基的主宾包合以及pABCD带正电荷的氨基与磷酸阴离子基的相互作用,pABCD对三磷酸腺苷的结合能力较强。FcA作为电活性探针可包含在pABCD腔内,产生较低的氧化峰电流。当ATP加入到pABCDFcA体系中时,pABCD空腔中的羧酸二茂铁被ATP置换,产生增强的氧化峰电流,并且氧化峰电流随ATP的浓度呈线性变化。该传感器选择性好,灵敏度高,线性范围为3.12×10
胆固醇是人体细胞和组织中的重要成分,在细胞膜的构建中起着非常重要的作用,也是胆汁酸、维生素D和甾体激素等的生物合成前体。Yang等[48]利用环糊精/聚乙酰苯胺/石墨烯修饰电极,构建了基于βCD与信号探针亚甲基蓝(Methylene blue,MB)和目标分子胆固醇的竞争性主客体识别的电化学传感器,用于选择性检测胆固醇。由于主客体识别作用,MB分子可进入βCD的疏水空腔,产生阳极峰电流。当胆固醇存在时,它可与βCD发生竞争性相互作用,从而取代MB分子,导致氧化峰电流的降低。该传感器检测范围为1~50 μmol/L,检出限为0.5 μmol/L,可用于血清中胆固醇的检测。
Yang等[49]设计了一种基于对磺化杯芳烃(pSulfonated calix[8]arene,SCX8)与信号探针/目标分子之间的竞争主客体识别的双信号电化学传感器,应用于乌头碱检测(图6)。该传感器分别利用亚甲基蓝(MB)和乌头碱作为探针和靶分子。由于主客体识别作用,MB分子可进入SCX8的疏水内腔,修饰后的SCX8/SWCNHs电极可产生明显阳极峰。在乌头碱存在时,它与SCX8发生竞争性相互作用取代MB分子,导致MB的氧化峰电流降低和乌头碱的氧化峰的出现。该电化学传感器对乌头碱的线性响应范围为1.00~10.00 μmol/L,检出限为0.18 μmol/L。
3.4金属离子的检测
金属离子特别是重金属离子对生物系统造成严重的环境污染和毒性。例如, Hg2+是一种高毒性的金属污染物,可引发多种急慢性疾病(例如心血管疾病、肾脏损害、神经和免疫系统紊乱)[54]。金属离子检测对环境保护和疾病预防具有重要意义。基于冠醚等超分子化合物对碱金属阳离子选择性识别和主客体作用,可实现对金属离子的特异性检测[55~63]。
Hu等[55]基于宿主客体识别和THg2+T的特异性相互作用,构建了电致发光(ECL)生物传感器,应用于Hg2+的检测(图 7)。他们将β环糊精修饰的钯纳米粒子(βCyclodextrinPd nanoparticles, βCYPdNPs)凝胶以及Ru(bpy)2+3层层组装到玻碳电极作为传感平台,同时将一端带Fc的DNA发卡探针通过βcyclodextrin(βCY)与二茂铁的主客体识别连接到βCYPdNPs上。当Hg2+不存在时,二茂铁可猝灭Ru(bpy)2+3的ECL信号。当Hg2+存在时,特异性的THg2+T相互作用使DNA发卡探针构象发生改变,二茂铁脱离β环糊精并且远离电极表面,导致ECL信号增强。该ECL生物传感器的线性响应范围为0.003~600 ng/mL,检出限为0.0015 ng/mL。
基于冠醚等大分子修饰的不对称纳米孔的电化学传感器可用于碱金属离子的检测[56~62]。PérezMitta等[59]利用冠醚对单个锥形纳米孔进行修饰,构建了可检测跨膜离子电流的纳米器件。纳米孔壁上发生的主客体离子识别过程可改变纳米孔的静电特性和整流特性。他们利用主客体化学,结合纳米流体元素,模拟特定生物通道的离子传输特性和门控功能。冠醚功能化纳米孔可特异性地与钾离子和钠离子结合,导致孔外电子读数发生显著变化[60]。Ali等[61,62]将纳米流体装置和杯冠醚功能化纳米孔相结合,用于检测铯离子和锂离子。
Yao等[63]构建了基于水溶性柱状芳烃(Watersoluble pillar[5]arene,WP5)和水溶性季铵盐二亚胺衍生物(Watersoluble quaternized perylene diimide derivative,G)超分子主客体体系(WP5G)的电化学传感器,可选择性检测Fe3+。G在水溶液中可形成具有强荧光的不规则团聚体。当加入WP5时,WP5(供体)和G(受体)发生光致电子转移(Photoinduced electron transfer,PET)反应,导致G荧光猝灭。当Fe3+存在时,Fe3+与柱芳烃的主客体识别可阻断超分子PET反应,导致G荧光恢复。该传感器可特异性检测Fe3+,检出限为2.13 ×10
3.5异构体的检测
大多数活性物质都具有手性,手性对映体的性能对生物活性、毒性、转运过程和代谢途径等方面的影响存在较大差异。通常情况下,只有一种异构体表现出完美的活性,而另一种可能无活性甚至会引起严重的副作用[64~66]。手性识别一直是化学和生物领域的研究热点。大多数主体分子对不同的异构体具有不同的结合力,因而可利用主客体作用对异构体进行选择性检测[67~71]。
Yi等[67]利用还原石墨烯包裹的碳纳米管(Carbon nanotubes wrapped with reduced graphene,CNTs@rGO)的协同作用和βCD与不同的目标物结合亲和力的不同,构建了双信号电化学传感器,可用于手性识别苯丙氨酸对映体(图8)。该传感器利用罗丹明B(RhB)和苯丙氨酸对映体(D苯丙氨酸和L苯丙氨酸)作为电化学指示剂。由于主客体识别作用,RhB可进入βCD的空腔,产生氧化峰电流。由于L苯丙氨酸(LPhenylalanine)和βCD之间具有较强的亲和力,L苯丙氨酸(LPhenylalanine)可与βCD空腔内的RhB发生竞争反应,导致RhB峰电流减少和L苯丙氨酸峰值电流的出现。这两个电化学信号的变化与L苯丙氨酸的浓度呈线性关系。但是,D苯丙氨酸和βCD之间的亲和力较弱,D苯丙氨酸不能取代RhB,因而不会发生RhB峰值电流变化。该双信号电化学传感器在0.05~22.0 μmol/L范围内对外消旋混合物中L苯丙氨酸对映体有较好的线性响应,检出限为0.013 μmol/L,同时比单信号传感器具有更宽的线性范围和更低的检出限。
Liang等[68]基于三维石墨烯与羟丙基β环糊精(Hydroxypropylβcyclodextrin,HPβCD)的耦合,利用HPβCD作为超分子手性识别元件以及三维石墨烯(3DG)作为电化学指示剂,构建了基于差分脉冲伏安法(DPV)的电化学传感器,用于识别色氨酸对映体。HPβCD对D色氨酸(DTrp)和L色氨酸(LTrp)具有不同主客体识别作用,其对靶L色氨酸结合亲和力较高,可用于色氨酸异构体检测。该传感器对这两种对映体的线性响应范围均为0.5~175 μmol/L,对LTrp和DTrp的检出限分别为9.6 nmol/L和38 nmol/L。
Yang等[69]集成主客体识别作用和内部滤波效应,构建了基于荧光金纳米团簇的传感器阵列(Gold nanoclusters,AuNCs),可用于識别硝基苯酚异构体。该传感器阵列装配3个不同的配体和βCD, 以及3个不同荧光发射的金纳米团簇,通过对3种硝基苯酚同分异构体荧光猝灭模式的线性判别分析,可对3种硝基苯酚同分异构体进行快速鉴别。该传感器阵列对于对硝基苯酚的线性检测范围为1~50 μmol/L, 检出限为0.21 μmol/L。另外,该传感器阵列不仅具有单一异构体识别能力,而且可区分邻硝基苯酚和对硝基苯酚两种异构体。
4总结与展望
综上所述,基于主客体作用的生物传感器可利用冠醚、环糊精和杯芳烃等主体分子对蛋白质[2336]、核糖核酸 [37~44]、生物小分子[45~53]、金属离子[55~63]和异构体[67~71]进行选择性检测,展示了广阔应用前景,但仍有许多问题有待解决。尤其是,基于主客体作用的传感器在生物分子的识别应用中多为单点识别。后续研究应侧重以下几个方面: (1)结合生物活性分子的特性构造相应的主体化合物,实现传感器固定化、器件化和传感器的重复利用,进一步实现分析检测过程的简便化与绿色化; (2)通过修饰功能性官能团或利用主体间协同作用构建混合主体,为客体提供更多识别位点,实现多重识别; (3)由超分子主体化合物设计合成生物模拟化合物(如蛋白质模拟化合物),这些化合物具有类似于抗原或酶的基质选择性识别机制,不仅稳定,而且价格低,可进一步用于研究其与核酸和蛋白质的相互作用,有望促进生物体内生理过程的研究。未来对分子识别的研究应向更高效和更广泛应用的方向发展。
References
1Yu G C, Jie K C, Huang F H. Chem. Rev., 2015, 115: 7240-7303
2YIN KaiLiang, XU RuiJun, XU YuanZhi, SUN XiaoQiang. Prog. Chem., 1997, 9(4): 337-348
殷开梁, 徐瑞钧, 徐元植, 孙小强. 化学进展, 1997, 9(4): 337-348
3Wang X N, Wang M W, Zhang Y Y, Miao X C, Huang Y Y, Zhang J, Sun L Z. Biosens. Bioelectron., 2016, 83: 91-96
4Sethuraman V, Muthuraja P, Raj J A, Manisankar P. Biosens. Bioelectron., 2016, 84: 112-119
5Xu Y Y, Liu L, Wang Z Y, Dai Z H. ACS Appl. Mater. Interfaces, 2016, 8(29): 18669-18674
6Yu H R, Hu J Q, Lu X H, Ju X J, Liu Z, Xie R. J. Phys Chem. B , 2015, 119(4): 1696-1705
7Szente L, Szemn J. Anal. Chem., 2013, 85(17): 8024-8030
8Prochowicz D, Kornowicz A, Lewinski J. Chem. Rev., 2017, 117(22): 13461-13501
9Espaol E, Maldonado M. Crit. Rev. Anal. Chem., 2019, 49(5): 383-394
10ZHANG Chun, ZHENG YanSong, MEI FuMing, LI GuangXing. Prog. Chem., 2004, 16(6): 934-939
張 春, 郑炎松, 梅付名, 李光兴. 化学进展, 2004, 16(6): 934-939
11Xu C H, Wang J C, Wan L, Lin J J, Wang X B. J. Mater. Chem., 2011, 21: 10463-10471
12Liu Z G, Xue Q, Guo Y J. Biosens. Bioelectron., 2017, 89: 444-452
13Tian X Q, Cheng C M, Yuan H Y, Du J, Xiao D, Xie S P, Choi M M F. Talanta, 2012, 93: 79-85
14Tang C, Qian Z, Huang Y, Xu J,Ao H, Zhao M, Feng H. Biosens. Bioelectron., 2016, 83: 274-280
15Li J, Yim D, Jang W D, Yoon J. Chem. Soc. Rev., 2017, 46(9): 2437-2458
16Quintana C, Suárez S, Hernández L. Sens. Actuators B, 2010, 149(1): 129-135
17Sun S, Hu X Y, Chen D, Shi J, Dong Y, Lin C, Wang L. Polym. Chem., 2013, 4(7): 2224-2229
18Wang W, Li Y, Sun M, Zhou C, Zhang Y, Li Y, Yang Q. Chem. Commun., 2012, 48(48): 6040-6042
19WU Meng, LIN ZhiHong, REN Shu. Chemical Sensor, 1997, 17(1): 292-299
吴 蒙, 林志红, 任 恕. 化学传感器, 1997, 17(1): 292-299
20XU FengBo, LI QingShan, ZHANG ZhengZhi, WU LiZhu. Photographic Science and Photochemistry, 2001, (3): 217-228
徐凤波, 李庆山, 张正之, 吴骊珠. 感光科学与光化学, 2001, (3): 217-228
21CHEN Zheng. Science, 1995, 47(01): 42-45
陈 政. 科学, 1995, 47(01): 42-45
22Zhu G, Wu L, Zhang X, Liu W, Zhang X, Chen J. ChemEur. J., 2013, 19(20): 6368-6373
23Xie S, Zhang J, Yuan Y, Chai Y Q, Yuan R. Chem. Commun., 2015, 51(16): 3387-3390
24Zhao M H, Cui L, Sun B, Wang Q B, Zhang C Y. Biosens. Bioelectron., 2020, 150: 111865
25Zhao M H, Cui L, Zhang, C Y. Chem. Commun., 2020, 56: 2971-2974
26Deng H H, Shi X Q, Peng H P, Zhuang Q Q, Yang Y, Liu A L, Chen W. ACS Appl. Mater. Interfaces, 2018, 10(6): 5358-5364
27Fan H, Li H, Wang Q, He P, Fang Y. Biosens. Bioelectron., 2012, 35(1): 33-36
28Wang X, Du D, Dong H, Song S, Koh K, Chen H. Biosens. Bioelectron., 2018, 99: 375-381
29Chen Q, Chen H, Zhao Y, Zhang F, Yang F, Tang J, He P. Biosens. Bioelectron., 2014, 54: 547-552
30Yang H, Wang H, Xiong C, Liu Y, Yuan R, Chai Y Q. Electrochim. Acta, 2016, 213: 512-519
31Shen W J, Zhuo Y, Chai Y Q, Yang Z H, Han J, Yuan R. ACS Appl. Mater. Interfaces, 2015, 7(7): 4127-4134
32Yu P, Zhang X, Zhou J, Xiong E, Li X, Chen J. Sci. Rep., 2015, 5: 16015
33Shang K, Wang X, Sun B, Cheng Z, Ai S. Biosens. Bioelectron., 2013, 45: 40-45
34Tan S, Han R, Wu S, Liang H, Zhao Y, Li C P. Talanta, 2019, 197: 130-137
35Gao J, Guo Z, Su F, Gao L, Peng X, Gao W, Wei Q. Biosens. Bioelectron., 2015, 63: 465-471
36Feng T, Qiao X, Wang H, Sun Z, Qi Y, Hong C. J. Mater. Chem. B, 2016, 4(5): 990-996
37Zheng J, Hu L, Zhang M, Xu J, He P. RSC Adv., 2015, 5(112): 92025-92032
38Yang S, Liu L, You M, Zhang F, Liao X, He P. Sensor. Actuators B, 2016, 227: 497-503
39Jiang J, Lin X, Ding D, Diao G. Biosens. Bioelectron., 2018, 114: 37-43
40Jiang J, Lin X, Diao G. ACS Appl. Mater. Interfaces, 2017, 9(42): 36688-36694
41Yang S, You M, Yang L, Zhang F, Wang Q, He P. J. Electroanal. Chem., 2016, 783: 161-166
42Fan H, Xing R, Xu Y, Wang Q, He P, Fang Y. Electrochem. Commun., 2010, 12(4): 501-504
43Abbaspour A, Noori A. Analyst, 2012, 137(8): 1860-1865
44Fan H, Xu Y, Chang Z, Xing R, Wang Q, He P, Fang Y. Biosens. Bioelectron., 2011, 26(5): 2655-2659
45Liu J, Leng X, Xiao Y, Hu C, Fu L. Nanoscale , 2015, 7(28): 11922-11927
46Wang L, Lei J, Ma R, Ju H. Anal. Chem., 2013, 85(13): 6505-6510
47Yang N, Wei Y, Kang X, Su Z. Anal. Methods, 2015, 7(24): 10129-10135
48Yang L, Zhao H, Fan S, Zhao G, Ran X, Li C P. RSC Adv., 2015, 5(79): 64146-64155
49Yang L, Ran X, Cai L, Li Y, Zhao H, Li C P. Biosens. Bioelectron., 2016, 83: 347-352
50Kingsford O J, Qian J, Zhang D, Yi Y, Zhu G. Anal. Methods, 2018, 10(45): 5372-5379
51Rahemi V, Vandamme J J, Garrido J M P J, Borges F, Brett C M A, Garrido E M P J. Talanta, 2012, 99: 288-293
52Abbaspour A, Noori A. Cyclodextrin A. Biosens. Bioelectron., 2011, 26(12): 4674-4680
53Chen H, Chen Q, Zhao Y, Zhang F, Yang F, Tang J, He P. Talanta, 2014, 121: 229-233
54Nolan E M, Lippard S J. Chem. Rev., 2008, 108: 3443-3480
55Hu Y, Liu Z, Zhan H, Shen Z. Anal. Methods, 2018, 10(7): 767-774
56Ali M, Nasir S, Ramirez P, Cervera J, Mafe S, Ensinger W. ACS Nano, 2012, 6: 9247-9257
57Ali M, Nasir S, Nguyen Q H, Sahoo J K, Tahir M N, Tremel W, Ensinger W. J. Am. Chem. Soc., 2011, 133: 17307-17314
58Han C, Su H, Sun Z, Wen L, Tian D, Xu K, Hu J, Wang A, Li H, Jiang L. Chem. Eur. J., 2013, 19: 9388-9395
59PérezMitta G, Albesa A G, Knoll W, Trautmann C, ToimilMolares M E, Azzaroni O. Nanoscale, 2015, 7(38): 15594-15598
60Liu Q, Xiao K, Wen L, Lu H, Liu Y, Kong X Y, Jiang L. J. Am. Chem. Soc., 2015, 137(37): 11976-11983
61Ali M, Ahmed I, Ramirez P, Nasir S, Cervera J, Mafe S, Ensinger W. Langmuir, 2017, 33(36): 9170-9177
62Ali M, Ahmed I, Ramirez P, Nasir S, Mafe S, Niemeyer C M, Ensinger W. Anal. Chem., 2018, 90(11): 6820-6826
63Yao Q, Lu B, Ji C, Cai Y, Yin M. ACS Appl. Mater. Interfaces, 2017, 9(41): 36320-36326
64Micali N, Engelkamp H, van Rhee P G, Christianen P C M, Scolaro L M, Maan J C. Nat. Chem., 2012, 4: 201-207
65Tiwari M P, Prasad A. Anal. Chim. Acta, 2015, 853: 1-18
66He Z, Zang S, Liu Y, He Y, Lei H. Biosens. Bioelectron., 2015, 73: 85-92.
67Yi Y, Zhang D, Ma Y, Wu X, Zhu G. Anal. Chem., 2019, 91(4): 2908-2915
68Liang W, Rong Y, Fan L, Dong W, Dong Q, Yang C, Wong W Y. J. Mater. Chem C, 2018, 6(47): 12822-12829
69Yang H, Lu F, Sun Y, Yuan Z, Lu C. Anal. Chem., 2018, 90(21): 12846-12853
70Zhu G, Gai P, Wu L, Zhang J, Zhang X, Chen J. ChemAsian J., 2012, 7(4): 732-737
71Wu S, Fan S, Tan S, Wang J, Li C P. RSC Adv., 2018, 8(2): 775-784
猜你喜欢
分析化学(2017年2期)2017-03-02
商业会计(2016年23期)2017-02-22
商场现代化(2016年27期)2017-02-14
企业技术开发·下旬刊(2016年11期)2016-12-27
现代经济信息(2016年24期)2016-11-09
商场现代化(2016年15期)2016-08-23
现代经济信息(2016年7期)2016-05-19
分析化学(2015年9期)2015-09-11
分析化学(2015年9期)2015-09-11
世纪桥(2015年2期)2015-04-03
