2019新型冠状病毒基因组的生物信息学分析
2020-07-09 01:27陈嘉源施劲松丘栋安阮吉寿
生物信息学 2020年2期
陈嘉源,施劲松,丘栋安,刘 畅,李 鑫,赵 强,阮吉寿,高 山*
(1.南开大学 生命科学学院, 天津300071; 2.东部战区总医院, 南京 210016;3.英国诺丁汉特伦特大学 生物科学系, 诺丁汉 NG11 8NS;4.南开大学 医学院, 天津300071;5.南开大学 数学科学学院, 天津 300071)
冠状病毒属(Coronavirus)的病毒是具有包膜的正链RNA病毒。由于2003年严重急性呼吸综合征(Severe Acute Respiratory Syndrome, SARS)和2012年中东呼吸综合征(Middle East Respiratory Syndrome, MERS)的爆发,冠状病毒逐渐成为病毒学领域的一个研究热点。2019年12月,中国武汉报道了冠状病毒引起的肺炎,其临床症状与2003年爆发的SARS不同,因此推断该病毒可能是冠状病毒的一个新变种。当前,冠状病毒溯源相对比较清晰:2003年,Enserink等在国际上首次提出了SARS冠状病毒(SARS Coronavirus, SARS-CoV)来自蝙蝠,而后通过果子狸传播给人类[1];再后来的病毒溯源研究都是根据蝙蝠-果子狸-人途径回溯,并重点研究各种蝙蝠体内的SARS样(SARS-like)冠状病毒与已知SARS冠状病毒的进化关系;特别是2013年,SARS冠状病毒(GenBank: AY274119.3)被溯源到中华菊头蝠(Rhinolophussinicus)[2]。尽管已有足够的冠状病毒基因组数据公开,有关冠状病毒跨物种传播和宿主适应性方面的研究却一直难以突破,特别是无法解释冠状病毒变异快、宿主多且具有较强的宿主适应性等特点。其中一个最主要原因就是冠状病毒基因组较大,多个基因功能未知。例如,SARS冠状病毒基因组(GenBank: AY274119.3)的全长是29 751 bp,比HIV-1病毒全长三倍还要多。另外,冠状病毒基因组注释中基因命名不统一、且大部分注释的蛋白质序列都来自预测,无实验验证,难以保证正确性。高通量测序的迅速发展,使大量冠状病毒基因组序列得以公开。然而,当前大部分的冠状病毒基因组研究都是简单使用全基因组或某个病毒结构基因的序列,后一种做法主观性较大。简单使用全基因组序列进行进化分析和重要功能或致病位点分析,不仅工作量巨大,而且大量与研究目的无关的基因组区域会对数据分析产生干扰。因此,不同于简单使用全基因组序列的其它研究,我们于2018年在国际上首次提出分子功能与进化分析相结合的研究思想,并应用于冠状病毒基因组的研究。分子功能与进化分析相结合,即仅使用与特定功能相关的基因组特征对一定变异范围内的序列进行进化分析[3]。这样不仅可以简化进化分析、提高信噪比,使分析结果更可靠,而且可以用于指导功能实验。2018年, 南开大学高山等在国际上首次报道了SARS冠状病毒中存在互补回文小RNA(complemented palindromic Small RNAs, cpsRNAs)的现象,并确定了Beta冠状病毒B亚群(BB冠状病毒)与功能相关的两个重要的基因组特征(Genomic feature)[3],即一段22 bp的互补回文序列(命名为Nankai complemented palindrome)与其所在的编码区(命名为Nankai CDS)。在上述研究中,对Nankai complemented palindrome进行突变分析的结果与使用Nankai CDS进行进化分析的结果一致支持SARS冠状病毒的跨物种传播途径是蝙蝠-果子狸-人。在分子功能与进化分析相结合的研究思想的指导下,本研究使用Nankai complemented palindrome和Nankai CDS对新发布的2019新型冠状病毒基因组(GenBank: MN908947)进行分析以期准确溯源,并对BB冠状病毒跨物种传播和宿主适应性进行初步研究。
1 数据与方法
在前期研究中,我们通过互补回文小RNA的发现从SARS冠状病毒MA-15株(GenBank: DQ497008)中确定了BB冠状病毒中与功能相关的两个重要的基因组特征,即一段22 bp的互补回文序列与其所在的编码区。这个编码区(DQ497008: 25689-26153)是BB冠状病毒基因组中一段高度保守的序列,在已知SARS冠状病毒基因组(如DQ497008)中只对应一个读码框ORF 3b;但在其它BB冠状病毒基因组中普遍对应多个读码框,因此ORF 3b无法代表其它BB冠状病毒基因组中的这段序列。本研究将22 bp的互补回文序列与其所在的编码区(可能是一个或多个读码框)分别命名为Nankai complemented palindrome和Nankai CDS。DQ497008和2003年爆发的SARS冠状病毒基因组(GenBank: AY274119.3)含有相同的Nankai complemented palindrome和Nankai CDS,因此在本研究中唯一代表SARS冠状病毒基因组。在前期研究中[3],我们的研究对象仅限于BB冠状病毒(本研究亦是如此),并进行如下操作[3]:提取NCBI GenBank数据库中全部BB冠状病毒的完整基因组(Complete genome)序列(以下简称病毒序列或序列);去除与Nankai CDS相似度在0.999以上的序列后,留下11条病毒序列(GenBank: JX993987、JX993988、GQ153539、GQ153540、GQ153542、DQ071615、DQ412042、DQ412043、AY515512、AY572034和DQ497008)用于进一步研究。本研究增加一条新发布的2019新型冠状病毒基因组的序列(GenBank: MN908947)和一条来自蝙蝠的序列(GenBank: MG772934),其Nankai CDS与2019新型冠状病毒Nankai CDS相似度最高(92.52%)。另外,由于本论文接收时2019新型冠状病毒基因组序列(GenBank: MN908947)尚未公开,实际使用的是来自互联网公开的序列(见致谢)。在本研究中,13条序列根据其宿主分为五组用于进一步研究,这五组命名为SARS(DQ497008)、果子狸Civet(AY515512和AY572034)、2019新型冠状病毒2019-nCoV(MN908947)、蝙蝠群体bat2(MG772934)和蝙蝠群体bat1(MG772934之外8条来自蝙蝠的序列)。在本研究中,进化分析使用软件PHYLIP v3.69,统计与作图使用软件R v2.15.3[4]。其它数据处理包括:病毒蛋白质预测和核苷酸三连子分析只考虑一个起始密码子ATG、三个终止密码子(TAA、TAG和TGA)和正向三个读码框;蛋白质预测不考虑小于20个氨基酸的蛋白质;最后,考虑四个起始密码子(ATG、GTG、CTG和TTG)和三个终止密码子重复蛋白质预测和核苷酸三连子分析以检验结果的可靠性(见结果)。
2 结 果
在经典定义中,DNA回文(DNA palindrome)和DNA互补回文(DNA complemented palindrome)序列都叫做DNA回文序列,而且大多数情况下,DNA回文序列的经典定义与我们新定义中的互补回文序列相同(例如ATCGCGAT)。根据我们的新定义[3],回文序列(DNA或RNA)要求序列(一条链)从5’到3’方向与从3’到5’方向的读取结果相同(例如ATCGGCTA)。回文和互补回文序列应该加以区别,主要基于三个原因:(1)我们的前期研究发现了回文和互补回文序列可能具有不同的生物学意义;(2)在动物病毒基因组中,回文和互补回文序列具有不同的分布特征;(3)经典定义是从DNA水平定义的,我们的定义考虑了单链DNA和RNA(特别针对病毒)情况。2004年,Chew等在国际上首次报道了SARS冠状病毒基因组中较短互补回文序列(新定义)的分布特征[5],其主要发现包括两点:(1)在所有分析的冠状病毒基因组中,长度为4 bp的互补回文序列出现频率显著较低;(2)长度为6 bp的互补回文序列仅在SARS冠状病毒(而不是所有冠状病毒)基因组中出现频率显著较低。因此,该研究的结论是SARS冠状病毒基因组含有较少的6 bp互补回文序列有可能利于该病毒有效规避宿主细胞内的某些防御机制而有利于病毒存活;该研究同时还发现了两个较长(14 bp以上)的互补回文序列分别是TCTTTAACAAGCTTGTTAAAGA和TAAAATTAATTTTA。但是,仅仅根据概率模型计算得到的此类回文或互补回文序列数量巨大,绝大部分无法与分子功能关联,因此没有进一步研究的价值。直到2018年,我们偶然发现了互补回文小RNA恰好来自这个22 bp的互补回文序列(命名为Nankai complemented palindrome),特别是发现了根据这个序列预测的RNA二级结构在BB冠状病毒基因组中高度保守(见图1A)。RNA水平的证据是将这个序列与分子功能建立关联的关键[3]。在前期研究中,我们对11个BB冠状病毒(见数据与方法)的Nankai complemented palindrome进行突变分析的结果与使用其所在的编码区(命名为Nankai CDS)进行进化分析的结果一致支持SARS冠状病毒的跨物种传播途径是蝙蝠-果子狸-人(见图1B红色方框内)[3]。
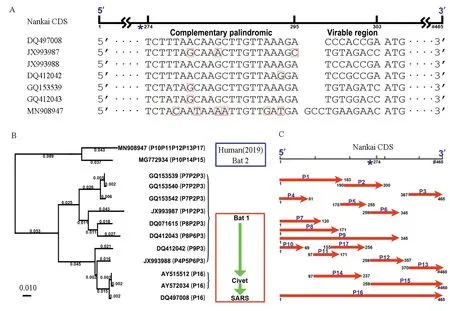
图1 2019新型冠状病毒的起源与可变翻译Fig.1 Origin and alternative translation of the 2019 novel coronavirus genome
注:A:Nankai CDS是BB冠状病毒基因组中一段高度保守的序列,包括一段22 bp的互补回文序列(用*表示)。Nankai CDS还包括一个8或11 bp的可变区,分别属于两种不同长度(是465或468 bp,用#表示)的Nankai CDS;B:进化树构建使用13条去除可变区的Nankai CDS。使用多种方法进行进化分析的结果一致,这里只展示邻接(Neighbor Joining,NJ)法的结果:2019新型冠状病毒源自蝙蝠某个群体(蓝色方框内),但与SARS冠状病毒差异巨大(红色方框内);用P1到P17(见图1C)可以对13个BB冠状病毒进行基因分型(小括号内),其结果与进化分析的结果一致。C:可变翻译导致从Nankai CDS可以预测出至少17个蛋白质,分别命名为P1到P17。这里展示的是使用一个起始密码子ATG和三个终止密码子进行蛋白质预测的结果,使用四个起始密码子(ATG、GTG、CTG和TTG)和三个终止密码子会预测更多的蛋白质,但不改变可变翻译相关结论。
Notes:A: The 22-bp complemented palindrome (named Nankai complemented palindrome) and the CDS (named Nankai CDS) have a high degree of evolutionary conservation in genomes of betacoronavirus subgroup B(BB coronavirus). An 8- or 11-bp long variable region in Nankai CDS resides in two types of Nankai CDS. The first type has a length of 465 bp, while the second type has a length of 468 bp.B: Consistent results were obtained by different phylogenetic analysis methods. Here, we only show the result using the NJ (Neighbor Joining) method. The 2019 novel coronavirus (in blue box) with large differences from the SARS coronavirus (in red box), may originate from BB coronaviruses in Chinese horseshoe bats. Phylogenetic analysis and genotyping (in parentheses) of 13 viruses using 17 putative proteins (Fig.1C) achieved consistent results. C: The putative proteins (named as P1 to P17) were predicted from the Nankai CDS using ATG as the start codon, and TAA, TAG and TGA as stop codons. The prediction using ATG, GTG, CTG and TTG as start codons, and TAA, TAG and TGA as stop codons did not change all conclusions in the present study.
在本研究中,所有13条Nankai CDS(见数据与方法)都含有一个完整的Nankai complemented palindrome和一个紧邻其3'端的长度为8或11 bp的可变区(Variable region)。使用MN908947(2019新型冠状病毒)中Nankai complemented palindrome预测的RNA二级结构仍然能形成稳定的茎环(Stem-loop)结构,再次验证了Nankai complemented palindrome在BB冠状病毒基因组中的保守性[3]。Nankai CDS中的可变区导致了长度变异(见图1A):MG772934(来自蝙蝠)和MN908947(2019新型冠状病毒)的Nankai CDS全长为468 bp(含11 bp可变区);其余11条序列的Nankai CDS全长为465 bp(含8 bp可变区)。由于无法确定每条Nankai CDS的可变区的变异方式,进行进化分析前,13条Nankai CDS去除可变区后即可对齐,长度统一到457 bp。使用多种方法(UPGMA、邻接法、最大简约法、最大似然法和贝叶斯法)对13条Nankai CDS进行进化分析的结果(见图1B)一致支持:(1)MN908947(2019新型冠状病毒)与MG772934(来自蝙蝠)在进化树的一个分支内(见图1B蓝色方框内),说明2019新型冠状病毒源自蝙蝠;(2)2019新型冠状病毒(见图1B蓝色方框内)与SARS冠状病毒(见图1B红色方框内)在不同分支,而且距离很大,这一结果与两者临床症状差异一致;(3)SARS冠状病毒的跨物种传播途径“蝙蝠-果子狸-人”(见图1B红色方框内)与2018年分析结果完全相同[3],证明了我们方法的可靠性。
Nankai CDS是BB冠状病毒基因组中一段高度保守的序列(见数据与方法),靠近其3'端的99个碱基(编码33个氨基酸)极端保守,不仅没有起始或终止密码子突变,也极少有其它突变,具有重要的研究和应用(如引物设计)价值。Nankai CDS在来自SARS和果子狸的beta冠状病毒基因组中是一个完整的读码框,编码蛋白P16(见图1C),但在来自其它宿主的冠状病毒基因组中(不包括3'端的99个碱基)出现了大量的起始或终止密码子突变(见表1),并导致Nankai CDS出现读码框的多样性,即可变翻译。由于去冗余后的完整基因组数据较少,需要从两个方面排除这些突变是来自测序错误等导致的假阳性。一方面,用13条Nankai CDS比对到NCBI NT数据库,看是否有其它冠状病毒序列支持这些突变。比对结果显示,大部分序列上的突变都能得到NT数据库中其它数据支持,只有三个例外:JX993988在Nankai CDS上的三个突变(C80A、T80A和T254A),MG772934(来自蝙蝠)和MN908947(2019新型冠状病毒)在Nankai CDS上共有的四个突变(T182A、C182A、T237A和T236G)没有其它数据支持。JX993988代表的病毒类型比较特殊,与其它来自蝙蝠的病毒差异较大(见图1B),这里不做进一步研究。另外一方面,统计13条Nankai CDS中核苷酸三连子出现次数,次数最高的前14位依次是:ATG(123次)、ACT(106次)、AAG(98次)、TTG(83次)、CAG(77次)、CAA(71次)、GTG(68次)、AAA(66次)、CTA(65次)、ACA(60次)、TTA(59次)、ATT(58次)、CGA(57次)和CTG(55次)。核苷酸三连子统计结果显示:前14位三连子出现次数之和超过三连子总数一半以上(1 046次/2 016次);6个三连子与起始密码子ATG只有0或1个核苷酸的差异;8个三连子与终止密码子(TAA、TAG和TGA)只有1个核苷酸的差异。统计结果说明了通过1个核苷酸突变(点突变)导致起始或终止密码子的丢失和获得的概率非常高。根据以上两个方面的分析结果,基本可以排除Nankai CDS中的大量起始或终止密码子突变是假阳性(见表1)。

表1 Nankai CDS内起始或终止密码子突变Table 1 Mutations in start or stop codons in Nankai CDS
注:可变翻译导致从Nankai CDS可以预测出至少17个蛋白质,分别命名为P1到P17(见图1C)。所有突变均来自17个预测的蛋白质,PP表示预测的蛋白质(Putative Protein)的名称;Start一列记录的是起始密码子的第一个碱基的位置;Stop一列记录的是终止密码子的最后一个碱基位置;Allele一列记录所在位置的密码子的全部类型;Mutation一列只记录导致获得起始或终止密码子的突变,突变格式为“突变前碱基-突变位置-突变后碱基”;AA表示PP的长度,单位是氨基酸(Amino Acid)。这里展示的是只使用一个起始密码子ATG和三个终止密码子进行蛋白质预测的结果;使用四个起始密码子(ATG、GTG、CTG和TTG)和三个终止密码子会预测更多的蛋白质,但不改变可变翻译相关结论。
Notes:The putative proteins (named as P1 to P17) were predicted from the Nankai CDS (Fig.1C). This table only includes the mutations in start or stop codons in 17 putative proteins. “PP” is Putative Protein; “Start” records the positions of the first nucleotides in start codons; “Stop” records the positions of the last nucleotides in stop codons; “Allele” records all types of codons on the positions; “Mutation” only records mutations which cause the acquisition of start or stop codons in the format “nucleotide before mutaion-position-nucleotide after mutaion”; “AA” is Amino Acid. The putative proteins were predicted from the Nankai CDS using ATG as the start codon, and TAA, TAG and TGA as stop codons. The prediction using ATG, GTG, CTG and TTG as start codons, and TAA, TAG and TGA as stop codons did not change all conclusions in the present study.
可变翻译导致从Nankai CDS可以预测出至少17个蛋白质(Putative Protein),分别命名为P1到P17(见图1C)。用P1到P17可以对13个病毒进行基因分型(基于可变翻译的基因分型),例如,MN908947(2019新型冠状病毒)可以表示为P10P11P12P13P17。我们发现基因分型相同或相近的病毒都在进化树上的同一个分支内(见图1B)。例如,bat1分支的病毒的基因分型都由三个蛋白质组成;SARS和果子狸分支都是P16;MN908947(2019新型冠状病毒)和MG772934(来自蝙蝠)都有P10。根据以上事实,可以得到以下推论:(1)BB冠状病毒(不限于Nankai CDS内)存在大量的可变翻译;(2)BB冠状病毒变异快、多样性高,仅仅13个病毒即可得到多达9种基因分型(见图1BC);(3)在SARS冠状病毒从蝙蝠到人传播过程中,Nankai CDS内多个点突变积累才形成P16;(4)在2019新型冠状病毒从蝙蝠到人传播过程中,P15只需一个点突变(C355T)即形成P12和P13,P14只需两个点突变(T170A和T191C)即形成P11和P17;(5)可变翻译可用于(但不限于)BB冠状病毒的基因分型,用P1到P17进行基因分型的结果与用Nankai CDS进行进化分析的结果一致。(6)BB冠状病毒可能通过可变翻译以适应不同宿主。
3 结 论
本研究证明了我们提出的分子功能与进化分析相结合的研究思想的可行性与可靠性,对生物大分子的功能或进化研究具有一定的指导意义,并对病毒或细菌等病原体的研究提供了新的路线和方向。因此,本研究除了解决2019新型冠状病毒溯源等具体问题,更主要贡献是提供了研究思想和方法。
1)使用Nankai CDS溯源分析的结果支持2019新型冠状病毒源自蝙蝠,但与SARS冠状病毒差异巨大;
2)从BB冠状病毒可变翻译中获取的信息可应用于其快速检测、基因分型、疫苗开发以及药物设计;
3)BB冠状病毒可能通过可变翻译以适应不同宿主;
4)对BB冠状病毒可变翻译的研究将有助于研究该病毒的变异、宿主适应性、感染以及致病机制等问题。
4 讨 论
病毒或细菌等原核生物的可变翻译现象很早就已经发现,但是其生物学意义尚未清楚,特别是缺乏基于较大数据的实证研究。我们在前期研究植物病毒[6]、昆虫病毒[7]、人病毒[8]、哺乳动物病毒(特别是非洲猪瘟病毒[9])和布鲁氏菌[10]的过程中,都没有明显观察到可变翻译现象,特别是BB冠状病毒Nankai CDS中这样大量而集中存在的可变翻译(仅仅13个病毒即可得到多达9种基因分型)极其罕见。此次实证研究结果达到了基于可变翻译的基因分型与进化分析的一致性,是一次较大突破,主要得益于以下几点:(1)分子功能与进化分析相结合的研究思想得以应用;(2)有蝙蝠-果子狸-人这一跨物种传播途径作为方向性指导;(3)无意中发现Nankai complemented palindrome和Nankai CDS;(4)来自多种宿主(蝙蝠、果子狸和人)的病毒数据,特别是阳性样本(如来自SARS和武汉肺炎患者)数据的积累。经典理论认为病毒与宿主共用一套翻译系统,然而病毒的蛋白质翻译也有其特殊性,导致本研究从Nankai CDS中预测的17种蛋白质不能提供准确的序列。蛋白质序列的准确度不影响本研究的所有结论和推论,而且为下一步蛋白质、细胞和动物水平的实验指出方向。
基于可变翻译的基因分型相同的病毒都在进化树上的同一个分支(同种宿主)内预示了beta冠状病毒可能通过可变翻译以适应不同宿主(见图1B)。但是,由于目前能获得的病毒数据仅仅来自少量宿主(蝙蝠、果子狸和人),这一推断的验证还需要积累来自更多种类宿主的病毒的数据。我们推测,病毒通过可变翻译以适应不同宿主作为一种普遍机制,并不限于冠状病毒或原核生物;真核生物中某些基因的可变剪接可能也有相似或相近的生物学功能。
BB冠状病毒通过可变翻译产生的多种蛋白质的致病性研究是一个更为重要的课题。当前公开的基因组数据虽然总量很大,但是,实际有效的阳性样本(有实验或临床记录证明其致病性)种类不足和缺乏阴性(临床实验确认的非SARS病毒)样本。致病性最明确的2019新型冠状病毒和SARS冠状病毒在Nankai CDS上各产生五个小蛋白和一个大蛋白(见图1C),并没有显示出蛋白质某个属性(如大小)与致病性的关系。在预测的17个蛋白质中,只有P16(长度为155个氨基酸)和P9(长度为115个氨基酸)是大蛋白质(见表1);P16(来自SARS冠状病毒)的致病性已知,而P9(来自蝙蝠的冠状病毒)未知。因此,来自蝙蝠的其它冠状病毒产生的蛋白(如P9)未必没有致病性,或许也具有导致2019肺炎或SARS的能力。本研究预测的17种蛋白质大小适中而且差异明显,可以很容易通过蛋白质组或外源表达等技术进一步验证其致病性。
致谢:感谢新型冠状病毒相关工作人员(省疾控中心等)的基础性工作,特别复旦大学张永振等公开的2019新型冠状病毒基因组数据,感谢南开大学生命科学学院动物系卜文俊、张涛、黄大卫、刘燕强和贺秉军等各位老师对我们生物信息学研究的长期支持。
猜你喜欢
生物学通报(2020年11期)2020-10-22
电脑报(2020年20期)2020-06-30
发明与创新·中学生(2019年6期)2019-06-26
中成药(2018年7期)2018-08-04
东方教育(2018年6期)2018-04-19
小溪流(画刊)(2016年12期)2017-02-04
微型小说选刊(2015年5期)2015-06-05
故事会(2015年18期)2015-05-14
小学生·多元智能大王(2014年5期)2014-07-24
中国烟草学报(2012年2期)2012-04-09