新型冠状病毒序列变异时空分析系统的设计与实现
2020-07-09 01:27:52李建勋徐德晨
生物信息学 2020年2期
李建勋,李 鑫,谢 康,徐德晨,李 杰
(哈尔滨工业大学 计算机科学与技术学院 生物信息学研究中心,哈尔滨 150001)
2019年12月出现以干咳、发热、浑身乏力等为典型症状的新型肺炎疫情,中国国家卫生健康委员会暂时将其命名为新型冠状病毒肺炎。2020年1月31日,世界卫生组织(WHO)宣布将新型冠状病毒肺炎列入“国际经济公共卫生事件”,并于2月11日将之命名为COVID-19[1]。同日,国际病毒分类委员会声明,将引发疾病的新型冠状病毒命名为SARS-CoV-2[2]。
1月份,新型冠状病毒肺炎扩散到国内多个省份和地区,后续世界各国也爆发了疫情[1]。截止到4月16日,中国累计确诊COVID-19共83 799例,但疫情已得到基本的控制,现存确诊1 891例,其中境外输入1 534例,占绝大部分[注]数据来源于国家及各省市地区卫健委,丁香医生整理(https://ncov.dxy.cn/ncovh5/view/pneumonia?)。然而,与此同时,新型冠状病毒已在世界多国相继爆发,全球累计确诊达2 047 104例,现存确诊140 675例,世界整体形势不容乐观。另外,国内外有多位相关领域的医学专家表示,新型冠状病毒短期内不会消失,可能与人类长期共存。
冠状病毒是一种包膜的单链RNA病毒,它的受体结合域能快速突变和重组,以适应并感染大量物种[3]。自病毒疫情爆发以来,学术界对于新型冠状病毒的基本传染数R0(Basic reproduction number)有许多研究和讨论,在多篇论文中给出的取值呈越来越大的趋势。4月7日,美国疾病管制局期刊《新兴传染病》发表了关于新型冠状病毒的最新研究结论[4],研究人员通过两种建模方法(到达模型和病例计数模型),最终计算得出新型冠状病毒肺炎R0中位数为5.7(95%CI 3.8-8.9),也就是说,一名新型冠状病毒肺炎患者可以传染5.7人,是之前认为的2~3倍。
在这种背景下,对新型冠状病毒序列的变异情况进行监控、分析及可视化,就显得尤为重要[5-6]。国内外已有一些相关的软件系统向用户开放,但是这些已有系统没有从时空角度进行分析的功能或相关功能仍有进一步完善的空间,例如北京志诺维思公司的“战新冠”系统(https://fight-sars2.genowis.com/),提供了对病毒序列的变异分析及可视化,但没有从时空角度进行分析的功能;国家生物信息中心开发的2019新型冠状病毒信息库线上系统(https://bigd.big.ac.cn/ncov/),提供了包含时空角度的病毒序列变异分析功能,但是其时空变异分析功能是使用热力图来实现,完全是静态图的形式,也不支持用户对基准序列、时间区间和地域范围进行选择,功能上仍然需要扩展和进一步完善。本文所设计和实现的新型冠状病毒变异时空分析系统用于解决这一问题,能够动态显示不同时间和地域病毒序列的变异境况(系统现已上线,网址http://bionet.org.cn/NCP-web/)。
1 系统的总体功能
新型冠状病毒变异时空分析系统的主要功能有三个:病毒序列来源统计、病毒序列变异统计及病毒序列比对。
1.1 系统首页
系统首页主要包括系统导航栏、系统信息简介、系统三个主要功能模块的简介。系统首页的部分截图如图1所示。
图1 系统首页截图展示Fig.1 Screenshot of the homepage
1.2 病毒序列来源统计
自新型冠状病毒爆发以来,各国研究人员在SARS-CoV-2测序方面做了大量的工作[7-12]。全球共享流感数据倡议组织(GISAID)将各国研究人员提交的病毒序列数据进行了整理和汇总,并在其官方网站(https://www.gisaid.org/)进行公开。本文使用的源数据来自GISAID官网[13]。
新型冠状病毒序列来源统计功能旨在对来自不同国家和地区的新型冠状病毒序列的数量进行统计、以地图和饼状图的形式进行可视化。
对新型冠状病毒序列来源的统计和可视化有两种形式——地图和饼状图(见图2)。

图2 地图形式的序列来源可视化Fig.2 Visualization of sequence sources in map form
图2展示了中国地图和世界地图上的新型冠状病毒序列来源统计,当用户将鼠标悬浮在目标国家或者地区上时,会以弹框的形式展示该国家或地区已提交的序列总数。
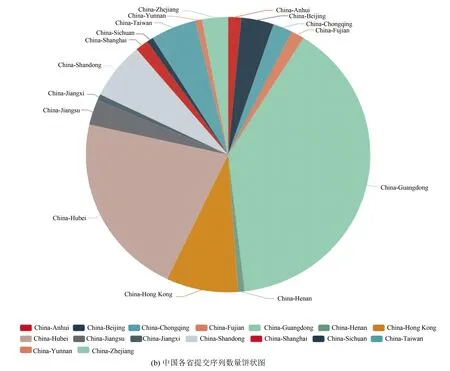
图3展示了饼状图形式下世界各国提交序列数量统计和中国各省提交序列数量统计。此外,系统还提供将以上二者结合的饼状图可视化。同时,当用户将鼠标悬浮在图中的扇形上时,系统会以弹框的形式展示该国家或省份提交的序列数占总数的百分比。
根据系统给出的结果(截止到3月19日的数据),可以发现世界范围内共提交SARS-CoV-2序列934条,中国提交的病毒序列最多,共267条,占28.59%;国内广东省提交的序列最多,共90条,占33.71%,湖北省次之,共42条,占15.73%。希望世界各国及时共享更多的SARS-CoV-2序列,促进COVID-19的研究。
1.3 病毒序列变异统计
系统将来自GISAID的序列数据进行筛选和预处理之后,将所有序列转换成等长的形式存入数据库(具体的筛选与预处理过程及方法见3.1节病毒序列数据的处理)。在此基础上,系统在第一次启动时,会比对数据库中所有序列,将每一个位点上频次最高的核苷酸作为该位点的标准核苷酸,从而得到一条统计意义上的标准序列(Standard sequence),后续可以此标准序列为基准进行变异分析(也支持由用户指定某一序列作为标准序列的变异分析)。
该模块下又分为序列变异统计分析和序列变异时空分析两个子功能模块。
1.3.1 序列变异统计分析
图4展示了系统为用户提供的选择入口,用户可在该入口下指定标准序列、选择待分析的目标序列集合、指定要分析的起始位点和终止位点。通过图4的设置面板完成分析基本参数选定并点击分析按钮,系统会给出病毒变异统计分析的两个结果,如图5和图6所示。

图4 选择基准序列与待分析序列Fig.4 Choice for base sequence and sequences to be analyzed

图5 在选择位点之间的变异病毒株数分布Fig.5 Distribution of variant virus strains between selected sites
图5展示了用户选定的目标序列集合与基准序列相比,具有不同变异位点数的病毒株数分布。除具有不同变异频数的病毒株数分布统计(见图5)之外,系统还会对病毒序列在不同时间和地区的平均变异率做统计(见图6)。该功能模块会将所有序列按照国家进行分组,每一组用不同的颜色加以区分,组内序列按照采样时间进行排序,给出所在地区在每一个采样日的平均变异率(每一个采样日内,对所在地区当天采样的所有病毒序列的变异率求取平均值)。该柱状图可以直观地展示某一地区内病毒序列的变异率随着时间递增的变化情况。
根据系统给出的分析结果(截止到3月13日的病毒序列数据),由图5可以发现,大部分序列的变异位点数少于10个,单条序列的最多变异数为31,有1条;由图6可以发现,中国在2020年1月29日采样的病毒序列的平均变异率最高,为0.09%,其余大部分序列变异率波动不大,绝大部分序列的变异率在0.04%以下。
1.3.2 序列变异时空分析
随着疫情的扩散,病毒在不同地区的变异率差异、在不同时间点的变异率变化这两个信息尤为重要,可以帮助科研人员对病毒进行更加精准的分析和研究,从而为疾病的研究和防治提供指导。该功能模块可以实现这一目标。
与病毒序列变异统计模块的参数面板相比,图7所示的参数面板具有更丰富的选项,包括标准序列、要分析的时间区间(以采样时间为准)、要分析的地域(以采样地域为准,可选择全世界的不同国家,也可以选择全国的不同省份)。在提交参数选项之后,系统会给出两个分析结果图,如图8和图9所示。

图7 变异时空分析的基本参数选项Fig.7 Basic parameter options for spatiotemporal analysis of variation
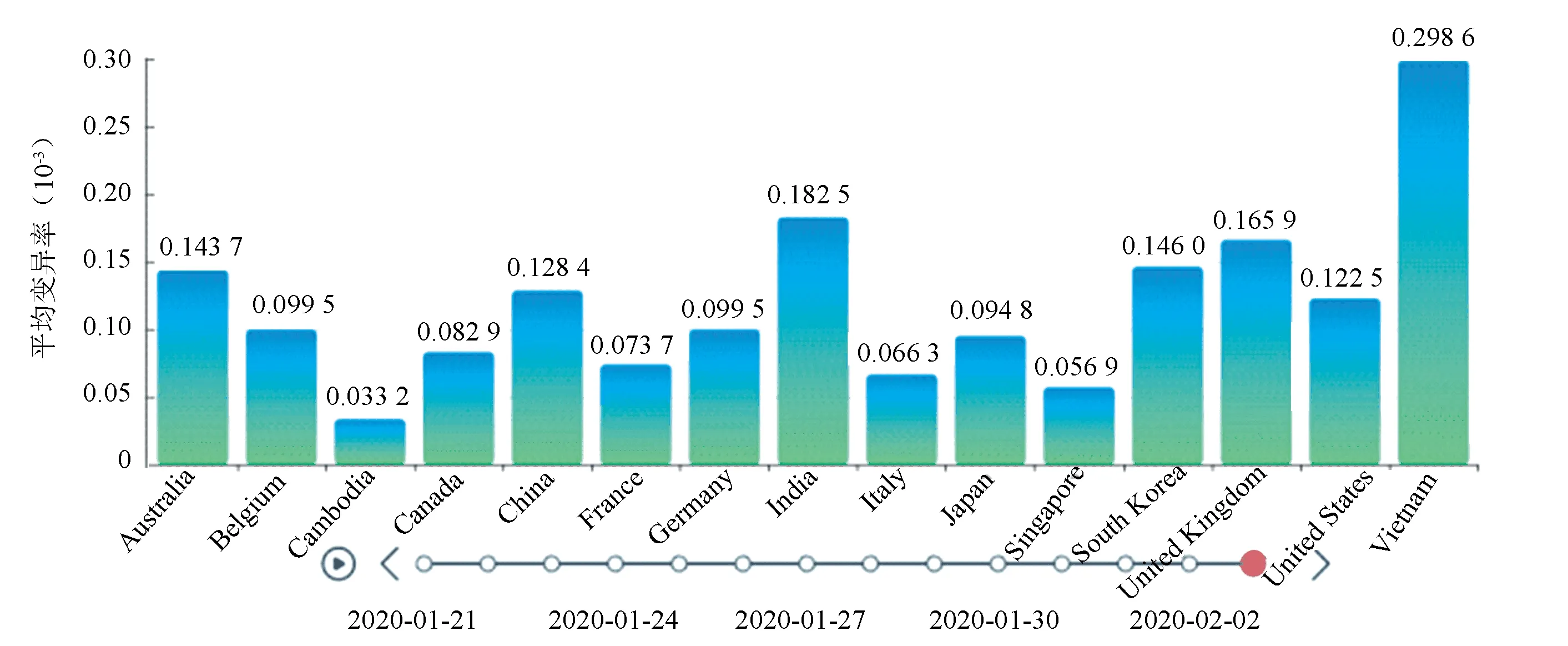
图8 不同时间下各地区的变异率Fig.8 Variation rates in different regions at different times
图8展示了在2020年1月21日到2020年2月3日之间十五个不同国家的病毒序列平均变异率(对所在国家或地区的截止到观测日期所采样的所有病毒序列的变异率求取平均值)的变化情况。在系统中实际操作时,该条形图是时间为变量进行动态播放的,在播放到最后一个时间节点时就会停止轮播。根据图8我们可以发现,在1月21日到2月3日之间,中国的平均变异率为0.013%,大致在平均水平左右;平均变异率最高的国家是越南,为0.030%;平均变异率最低的国家是柬埔寨,为0.003%。
图9展示的是序列变异时空分析模块给出的第二个分析结果。与图8相同,该折线图在实际操作时也是随时间变化动态播放。该图展示的是1月21日到2月3日之间,十五个不同国家整体的病毒序列变异情况。其中黄色折线为最大变异率、绿色折线为最小变异率、蓝色折线为平均变异率(对全球所有国家或者国内所有省份的截止到观测日期所采样的所有病毒序列的变异率求取平均值),鼠标在图上悬浮时会给出详细信息。根据图9可以发现,1月21日到2月3日之间,病毒序列的最大变异率为0.09%,结合1.3.1节的图6,可知该序列在1月29日采样于中国广东;最小变异率为0%,该序列在1月23日被采样;平均变异率在0.01%左右波动。
1.4 病毒序列比对
除病毒序列的来源统计和病毒序列变异统计两个模块外,系统还设有病毒序列比对模块。该模块下又细分为病毒序列基本信息展示和病毒序列差异比对两个子模块。
1.4.1 病毒序列基本信息展示
GISAID官网除了提供病毒序列之外,还给出了一些病毒序列相关的信息,其中包括病毒序列号、病毒名称、采样地点、采样实验室、提交实验室、作者信息、采样时间,系统以表格的形式对这些信息做了展示(见图10)。表格的每一行对应一个病毒序列,所有病毒序列按照国家分组并按照采样时间升序排序。同时,在实际操作系统时,表中每一行前面都有一个单选框,用户在感兴趣的单选框内打钩,即可选中该行所对应的序列,进行病毒序列差异比对。

图9 不同时间下所有地区的累计变异率Fig.9 Cumulative variation of all regions at different times

图10 序列的基本信息表Fig.10 Sequence basic information table
1.4.2 病毒序列差异比对
图11展示的是系统对EPI_ISL_403932、EPI_ISL_408484、EPI_ISL_410984、 EPI_ISL_415462 和EPI_ISL_415652共五条序列在1到30145这一位点区间上进行差异比对的结果,最终以表格的形式将所有存在差异的位点一一列出。根据表格可以发现,上述五条序列在298、1 247、3 094、8 839、9 495、11 143、13 111、14 468、15 384、19 251、23 467、24 098、24 926、26 793、28 141、28 208、29 159这些位点上存在差异,即这些位点都是可能发生了变异的位点。

图11 病毒序列差异比对Fig.11 Virus sequence difference alignment
2 系统的设计与实现
2.1 数据库
系统使用了关系型数据库MySQL,以每一条序列作为一个实体,将所有数据都整合到一张表中。即表中综合了每个序列的基本信息(序列名、病毒名、采样地点、采样时间和提交时间等)和核苷酸排布信息(以字符串形式存储,在数据库中字段的类型为不限长度的text类型)。表的主要字段如图12所示。

图12 表格主要字段Fig.12 Main fields of the data table
2.2 服务端
系统服务器端使用Spring、SpringMVC配合MyBatis框架作为主要的技术进行实现[14],并将项目部署在阿里云ECS服务器上提供服务。
2.3 客户端
系统的客户端主要采用jquery、bootstrap、echarts框架[15]进行开发。jquery框架作为一个快速简洁的轻量型框架,提供了完备的功能接口,支持丰富的插件便于扩展,同时能够较好的支持服务端与客户端得数据交互。bootstrap框架具有响应式布局设计,能让网站兼容不同分辨率设备,给用户较好的体验。而echarts框架能够将我们的数据以图表可视化的方式进行呈现,有利于用户对数据进行阅读和分析。
3 数据处理与分析
3.1 病毒序列数据的处理
为了寻找发生变异的位点,我们首先将新型冠状病毒序列进行了多重序列比对。本研究中使用的多序列对比工具是MUSCLE[16],软件版本为3.8.31。比起其它常用的序列对比工具,如ClustalW[17]、T-coffee[18],MUSCLE在保证了较高的准确度的同时,具有更快的多序列比对速度。在参数选择上,我们设置了参数-diags来适用于高度同源的新型冠状病毒序列,同时最大迭代次数(-maxiters)选择16来保证多序列对比的准确性,而其它参数选择了默认设置。
多重序列比对之后,多个新型冠状病毒序列间同源序列的相同核苷酸将处在相同的位点上,而如果原始序列在该位点中不存在核苷酸,将用字符“-”来表示。经过多重序列比对的序列都将拥有相同的序列长度。
对于对比后的序列,根据编码区异常字符(N)的数量以及异常缺失位点数量,我们设置30个为病毒序列的筛选条件,异常位点大于筛选条件值的序列将被遗弃。
将筛选后的比对结果转化为核苷酸矩阵,每条序列为行,每个位点为列,寻找每一列是否有超过一种核苷酸,如果结果为是,则该位点为有变异。
经如上方法对来自GISAID的数据进行预处理之后,即可将所有病毒序列转换为等长的形式,在此基础上即可按照2.1节所述内容进行数据的整合和数据库表的设计与填充。
3.2 病毒变异的时空分析
随着时间线的延长,各国提交的序列也逐渐增多,相应的各地区病毒序列的平均变异率也在逐渐增加,如图9所示。这也符合包膜的阳性单链RNA病毒在传播过程中容易变异的特点。我们也基于图8所对应的功能模块对截止到3月13日的不同国家或地区的平均变异率做了比较分析,找出了平均变异率较高和较低的部分国家以及国内部分省份,分别如表1和表2所示。可以看出,到目前为止,病毒序列的平均变异率处于一个较小的波动范围内,病毒仍然处于较为稳定的状态。
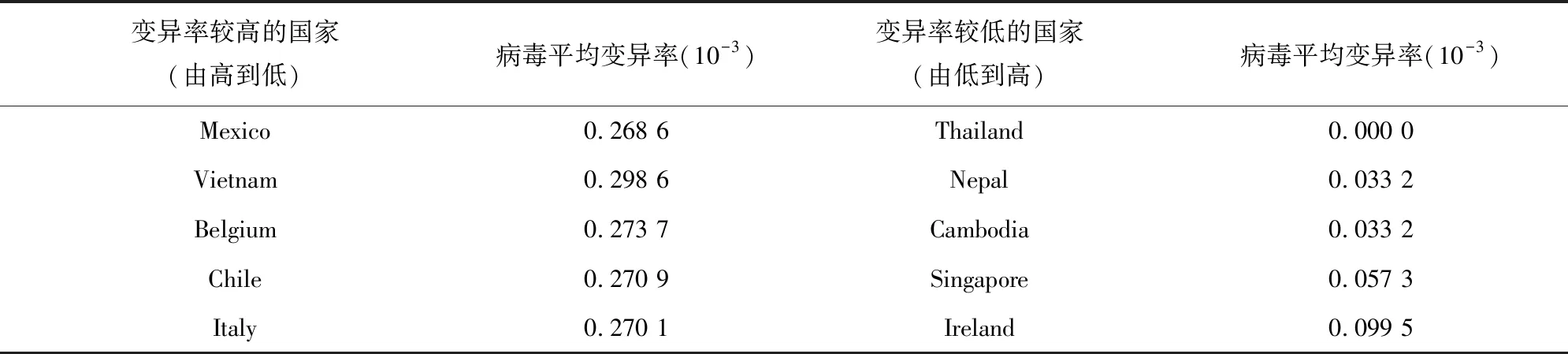
表1 病毒变异率较高和较低的部分国家Table 1 Countries with higher and lower virus variation rates

表2 病毒变异率较高和较低的中国部分省份Table 2 Provinces in China with higher and lower virus variation rates
结合在选择位点之间的变异病毒株数分析结果(图5所对应的功能模块),大部分有变异发生的病毒序列的变异位点数量小于12个,这也印证了目前病毒变异率较小这一结果。
进一步比较不同地区不同时间的病毒变异率,发现随着地区与时间的不同,病毒平均变异率也有差异,但总体仍处于在较小范围内波动的状态。
对于病毒序列变异分析,用户可以选择感兴趣的起始位点和终止位点对序列进行分析,比如针对病毒的S蛋白编码区,在本次多重序列对比中对应起始与终止位置分别为21 627和25 448,在系统中选择后,可以看到变异数量的信息,其中变异位点数量(SNP)为0和1的病毒数量最多,对应病毒株数分别为302和194,其余SNP数分别为2、3和7,对应病毒株数分别为28、6和1,大部分病毒序列均未发生明显改变。
4 总结
本系统对来源于全球共享流感数据倡议组织(GISAID)的新型冠状病毒数据进行整合与分析,主要实现了以下功能:
1)对数据的来源进行可视化分析。
2)对序列的基本信息进行展示,并支持选定序列之间核苷酸比较。
3)以动态图的形式呈现不同时间下各地区的病毒变异率,以及所有地区的累计变异率,使用户能够直观感受病毒的时空变异情况。
4)对选择位点之间的变异病毒株数进行统计,并以柱状图的形式对病毒的变异率进行时间空间上的展示,方便用户对病毒的变异情况进行分析。
该系统以一种更为直观的方式呈现病毒的具体信息,同时以图表的方式,对病毒的变异情况进行了展示,方便用户对新型冠状病毒的序列进行分析,对新型冠状病毒防治的研究具有重要的参考意义。
猜你喜欢
四川党的建设(2022年8期)2022-04-28 21:29:35
上海金属(2021年6期)2021-12-02 10:47:20
昆明医科大学学报(2021年3期)2021-07-22 07:40:04
小学生学习指导(低年级)(2020年11期)2020-12-14 07:28:10
趣味(数学)(2020年4期)2020-07-27 01:44:16
支部建设(2020年15期)2020-07-08 12:34:32
生物学通报(2019年3期)2019-02-17 18:03:58
作文大王·低年级(2018年10期)2018-12-06 06:22:44
小猕猴智力画刊(2016年5期)2016-05-14 09:21:39
百科知识(2015年18期)2015-09-10 07:22:44