
基于VAE 的编码DNA 载体阻断事件聚类分析与研究
2020-07-08 07:35魏梓轩周家乐
华东理工大学学报(自然科学版) 2020年3期
魏梓轩, 周家乐
(华东理工大学信息科学与工程学院,上海 200237)
纳米孔道检测技术作为一种可用于单分子检测的有力工具,具有巨大的研究价值和应用潜力[1-4]。目前,纳米孔道已经被成功应用于DNA 测序[5]、蛋白质检测[6]、蛋白质折叠[7]等研究中。纳米孔道检测装置通常由纳米孔道及其连通的两部分电解质溶液组成,在外加电场的驱动下,待测物分子穿越纳米孔道时,使得流经纳米孔道的离子电流发生变化,进而形成特征阻断电流[8]。因此,待测物分子的穿越行为对离子电流产生的信号进行了调制,这些信号中包含了分子电性、尺寸和结构等特异性信息[3,9-10]。
待测物分子穿越纳米孔道产生的瞬时电流阻断为一次阻断事件。为识别这些事件,可以利用电流阈值的方法来实现[11]。然而,由于纳米孔道信号的信噪比较低,增加了后续特征提取和分析的困难。传统的数据分析方法主要提取事件持续时间和阻断幅值两个特征,通过研究其散点分布对待测物分子进行区分[12-13]。该方法往往依赖于纳米孔道对不同分析物的分辨率,具有一定的随机性。即使利用具有较高分子空间结构分辨率的纳米孔道,也难以完全对混合待测物散点分布中的每一个事件进行区分,因此,该数据分析方法对纳米孔道阻断事件的分辨率较低[14]。
文献[15]利用DNA 分子链作为蛋白质分子的载体,并在载体DNA 链上的不同位置加以哑铃型束夹修饰。镶嵌哑铃型束夹后,载体DNA 分子镶嵌部位的体积增大,在穿越孔道过程中会产生二级阻断事件。通过在预先设定的若干位置进行束夹镶嵌,形成编码结构,进一步通过分析二级阻断事件特征,便可对编码DNA 载体上的蛋白质分子进行分类。为实现对编码DNA 载体的纳米孔道信号进行分析,需精确识别二级阻断事件的特征。然而,由于纳米孔道检测存在的局限,如幅值变化、信噪比低、信号持续时间的分布范围大、重叠事件、DNA 分子链的缠绕和折叠等问题,使得自动化的分析方法难以适用于各种复杂的情况,需要依赖于手动调整参数[4,16-18]。Misiunas 等[10]利用卷积神经网络的自动特征提取特性提出了QuipuNet 网络结构,对编码DNA 载体种类和有无蛋白质绑定进行分类,取得了较高精度。然而,QuipuNet 作为一种监督分类方法,依赖于人工对阻断事件进行精确标注以用于模型的训练,这需要耗费大量的时间,效率低。无监督学习方法不依赖于对阻断事件进行标注,可自动发现信号中的类簇[19-20]。基于这一优势,本文建立了基于深度嵌入特征[21]和变分自编码器[22]的聚类模型,该模型实现了对不同持续时间的阻断事件的低维嵌入,并在嵌入特征上进行聚类。为验证此模型的聚类效果,将其与经典的聚类方法进行了对比分析。在无监督聚类精度的评价标准下,本文模型实现了对纳米孔道数据的精确分析。
1 编码DNA 载体阻断事件数据集描述
Misiunas 等[10]将编码DNA 载体的阻断事件的表格信息整理到HDF5 文件格式中,可用Pandas 提供的数据接口进行读取。该数据集提供了32 个纳米孔道产生的58 178 条数据,包含了“000”~“111”8 种编码DNA 载体有无蛋白质绑定的阻断事件信息。每条数据包含编码、纳米孔道编号、阻断事件信号数据点、有无蛋白质绑定、展开幅值等信息。本文采用该数据集,使用阻断事件信号数据点作为模型输入,输出得到对应信号的8 种编码聚类簇,并利用已有的标注信息对算法的效果进行分析。在对信号进行聚类前,还需对阻断事件进行预处理,利用载体DNA 分子链的展开幅值(Unfolded level)去除不同纳米孔道对信号幅值产生的影响,以避免模型对纳米孔道编号过拟合。
2 深度嵌入聚类与变分自编码器
2.1 深度嵌入聚类


2.2 变分自编码器


3 无监督聚类框架
3.1 基于变分自编码器的深度嵌入聚类



图1 基于变分自编码器的深度嵌入聚类Fig. 1 Deep embedded clustering based on the variational auto encoder

3.2 变分自编码器的网络结构设计
将卷积网络结构设计用于聚类框架的变分自编码器。如图2 所示,该网络输入维度为700,卷积层输出按两组相同维度的特征图为一个元胞进行链接,其维度按照自编码器的通用设计方法采用逐级降低方式进行设定。因此,设定卷积层输出的维度为{350, 350, 128, 128, 87, 87},那么卷积核的大小依次为{7, 7, 5, 5, 3, 3}。对于越深层的卷积层,选择更多数目的特征图来提取信号中的特征,其数目依次为{16, 16, 32, 32, 64, 64}。将卷积层输出连接至维度为1 024 的全连接层,并分别连接至均值μ 和标准差σ,其维度为32。再通过式(8)所示的重参数技巧,利用均值μ 和标准差σ 进行高斯分布的采样,得到隐变量表示z,至此构建成编码器结构。解码器按照与编码器镜像的方式进行构建,卷积算子利用反卷积算子进行代替,以还原出信号空间的维度。该网络中的激活函数全部采用LeakyRelu 函数:

其中:x 为各层的线性输出;γ 为待优化参数。
4 仿真及结果分析
4.1 数据预处理
纳米孔道检测技术存在的局限使得编码DNA 载体阻断事件中包含着与具体编码类别无关的干扰,因此,在应用本文的聚类方法前,有必要对数据进行预处理,以降低聚类模型对噪声特征的过拟合风险。由于纳米孔道的直径具有固定尺寸,编码DNA 载体自身直径和加之以靶位点后的直径之间存在较大的差距,因此会在孔道中产生不同的占位作用,从而产生不同大小的二级阻断电流幅值。图3示出了8 种不同编码DNA 载体对应的阻断电流信号,图中红色虚线表示载体DNA 分子链的展开幅值,黑色虚线表示阻断事件的开孔电流幅值,箭头表示开始位点至结束位点的方向。
根据图3 中箭头指向可以判断,编码DNA 载体两端进入纳米孔道的顺序是不固定的,但通过判断靶位点尖峰的偏向可以判断载体两端的进入顺序,进而从高位到低位读出编码。同一种类、不同批次的纳米孔道,由于制备条件的差异,会在尺寸和结构上存在偏差,尤其是固体纳米孔道。如图3 中红色虚线表示了编码DNA 载体进入纳米孔道时去折叠过程中的主要电流水平,是对电流数据设置3 个聚类中心进行聚类分析的结果。在将开孔电流对齐后,可看到红色虚线所示的电流水平存在明显差异,因此,需要对这些信号的去折叠电流水平进行对齐,以防止模型聚焦于孔差异而带来错误的聚类结果。

图2 聚类框架中的变分编码器结构Fig. 2 Structure of the encoder of variational auto encoder in the clustering framework
另外,从图3 中还可以发现原始阻断事件的持续时间并不一致,这是因为分子穿过纳米孔道过程中受到初始速度、分子结构、进入角度等随机因素影响。传统分析方法通过统计分布来分析待测碱基链长度[12,26],而本文将阻断事件的信号输入到变分自编码器中。由于变分自编码器为一个神经网络模型,输入信号维度固定,因此,除了将信号的电流水平对齐,还需要对信号进行长度补全。选取每个信号的前50 个数据点(开孔电流信号)分别计算其标准差,得到平均值为0.009 5。进而,选取均值μ = 0、标准差σ = 0.009 5 的高斯白噪声,对信号的末尾补全至总共含700 个数据点。最终,不同编码DNA 载体的阻断事件数量如表1 所示。

图3 8 种编码DNA 载体的阻断电流信号Fig. 3 Blockade signals of eight encoded DNA carriers
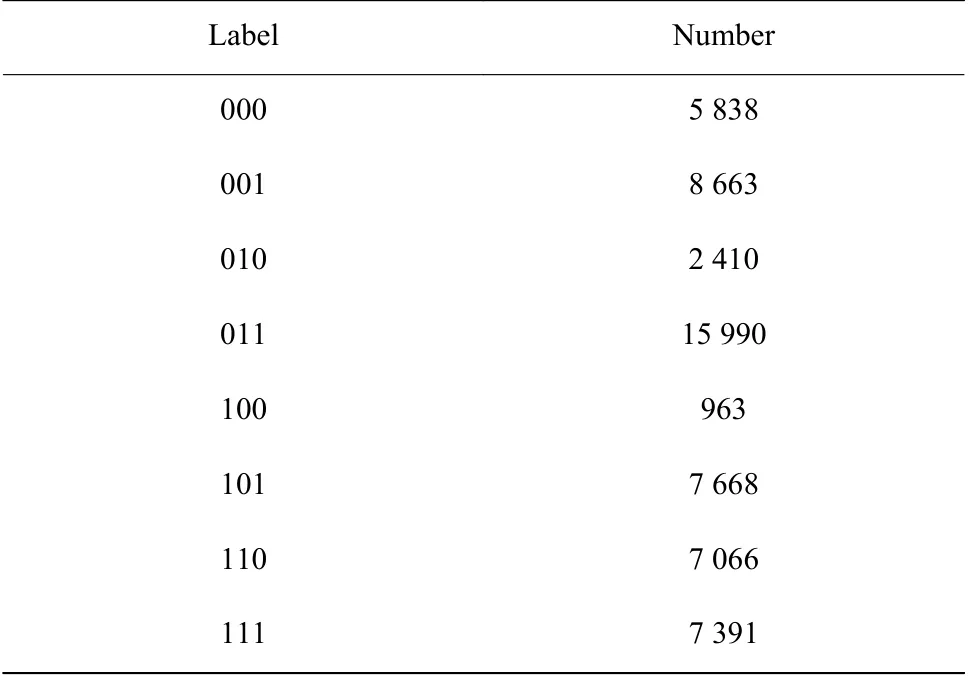
表1 编码DNA 载体的阻断事件数量Table 1 Numbers of blockade events produced by encoded DNA carriers
4.2 仿真结果及分析
仿真平台配置:Intel®Xeon®CPU E5-2650 v4 @2.20 GHz, 252 GB RAM, 64-bit GNU/Linux Centos 7,Python 3.6.8, Keras 2.2.4, Tensorflow-mkl 1.12.0。本文选择结合动量和自适应学习率的优化方法(Adam),对式(12)所示的损失函数进行优化。训练时,所选取的批大小为512,最大迭代次数为20 000,目标分布的更新间隔为140 次,即T = 140。
利用2.3 节中的优化方法和参数,对基于变分自编码器深度聚类框架进行优化。随机选取了4 个聚类中心,并分别绘制出离聚类中心最近的5 个阻断事件样本,如图4 所示。结合表1 中的数据样本数量可以发现,离聚类中心距离较近的样本中出现频率较高的“011”、“101”等编码,同时也是样本数据集中占比较高的编码。表1 中的数据存在严重不平衡的现象,表明聚类过程中占比较高的样本会完全淹没占比较小的样本,从而产生与编码无关的聚类模型。
对表1 中数据进行随机降采样,使得8 种编码的分布保持一致,最终得到每个编码对应样本数量为963 的数据集。在降采样后的数据集上,利用2.3 节的方法对上述模型重新训练。选取4 类聚类中心,并分别列出5 个最相似样本,如图5 所示。对比图5 中的结果可以发现,降采样后的聚类效果得到提升,每类聚类中心周围的样本纯度较高。因此,对原始的数据样本进行降采样后,训练得到的模型在聚类效果上得到了提升。
利用K-means、Auto-encoder + K-means (AE + Kmeans)、VAE + K-means、IDEC[24]、基于变分自编码器的IDEC(VAE + IDEC)分别对降采样后的数据进行聚类,利用式(13)对聚类后的结果进行评价,结果如表2 所示。不难发现,可聚类的变分自编码器模型相较于其他聚类算法可显著提升分析精度,提升量最高可达29%。K-means 聚类算法的结果较差,原因在于该算法要求原始数据严格对齐,更适用于表格型数据,并且聚类效果也容易受到噪声的干扰。而编码DNA 载体阻断事件信号具有时序数据中变形、位移等特征,这对K-means 中计算样本与聚类中心距离的方法提出了更高的要求。因此,变分自编码器能更有效地提取出原始信号中与阻断事件类型相关的特征,进而通过将变分自编码器与聚类层联合训练的方式,使得编码器产生的隐变量分布逐渐向聚类中心对齐。AE + K-means 等两阶段的聚类方法中,表示特征转换的编码层网络的训练与聚类过程是分离的,因此难以保证编码器所转换特征的对齐效果。在计算样本与聚类中心距离时,引入了一定的噪声,使得聚类效果较差。基于VAE 的聚类模型相比于基于AE 的模型都取得了较高的聚类精度,原因在于VAE 对数据的概率分布参数进行建模,而不仅仅是对输入数据进行压缩表示。概率分布参数反映了输入数据分布的本质特征,基于此可以得到更优的聚类精度。
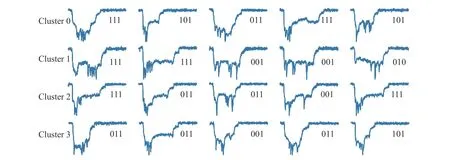
图4 0 ~ 3 类中离聚类中心最近的5 个样本Fig. 4 Five samples closest to the centroids of cluster 0 to 3
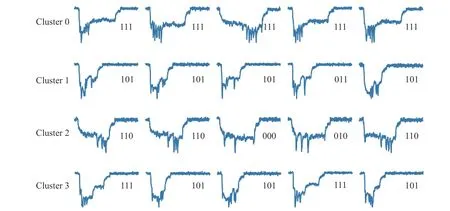
图5 降采样后的聚类结果Fig. 5 Clustering results after under-sampling
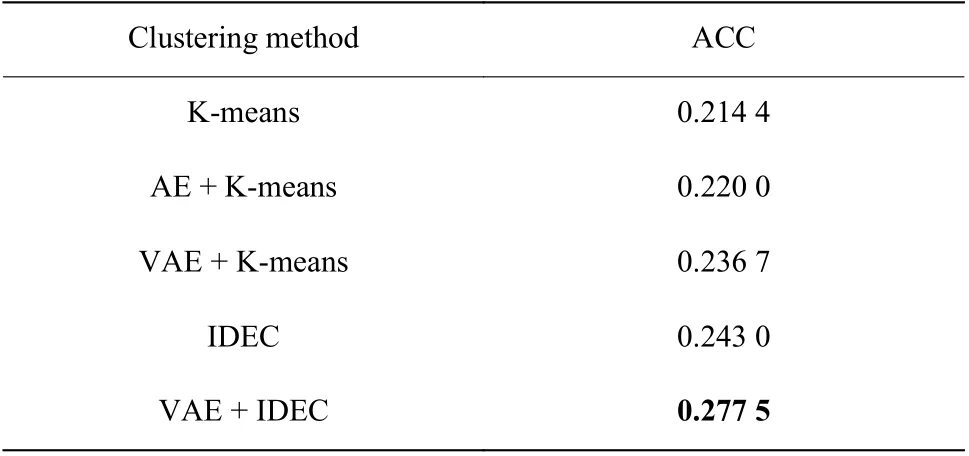
表2 K-means、AE + K-means、VAE + K-means、IDEC、VAE +IDEC 的聚类结果比较(8 个聚类中心)Table 2 Clustering results comparison of K-means, AE + Kmeans, VAE + K-means, IDEC, VAE + IDEC (8 clusters)
由于原始数据,即编码DNA 载体的阻断电流信号,存在不规则的信号特征,如持续时间分布的方差、极差较大,使得原本分布在时间域上的数据又可能分离出更多子类。因此,通过对表2 中各个聚类方法的比较,本文认为变分自编码的隐变量特征最大化地保留了信号特征,通过对隐变量进行分布假设,使得原始信号与类别相关的特征被转换到更低维的特征分布中。
从图3 可以发现,相同编码的DNA 载体进入纳米孔道的方向是不确定的。因此,编码001 和编码100、编码011 和编码110 的DNA 载体穿越纳米孔道会产生相同时序的阻断事件。在该聚类问题中,无论是传统聚类算法还是本文提出的基于变分自编码器的聚类算法,都无法直接利用尖峰的偏向信息来建立较优的聚类模型,所以本文将编码001 和编码100、编码011 和编码110 的数据分别设定为同一类别,得到6 个中心的聚类模型。聚类结果如表3 所示。
将上述结果与表2 的结果进行对比,可以发现所有聚类算法的无监督聚类精度都得到了一定的提升,总体上升了12%。同时,本文提出的基于变分自编码的深度嵌入聚类模型得到了较优的聚类精度,最高提升了23%。这说明基于变分自编码器隐变量模型对于信息感知和压缩的有效性。本文设计的变分自编码器模型采用了卷积神经网络的隐层结构,可见卷积运算在纳米孔道信号滤波或特征提取中的实用性。

表3 K-means、AE + K-means、VAE + K-means、IDEC、VAE +IDEC 的聚类结果比较(6 个聚类中心) Table 3 Clustering results comparison of K-means, AE + Kmeans, VAE + K-means, IDEC, VAE + IDEC (6 clusters)
5 结 论
数字编码的DNA 载体为检测蛋白质分子提供了重要的途径,然而传统的数据分析方法往往依赖于手动进行大量的调参。本文提出了一种基于变分自编码器的深度嵌入聚类方法。首先,利用阻断事件数据集对变分自编码器进行预训练,对信号的隐变量空间进行建模。再将预训练好的变分自编码器加入到深度嵌入聚类的框架中,在隐变量空间中对信号的嵌入特征进行聚类。隐变量模型能够保留信号中的主要特征,可以有效过滤阻断事件信号的噪声、变形等干扰。与传统的以及多阶段的聚类算法相比较,本文提出的聚类框架以及设计的变分自编码器网络实现了较高精度的聚类效果。
本文的无监督聚类方法与卷积神经网络模型在DNA 载体阻断事件中的高精度监督分类效果,展示了卷积运算在纳米孔道数据处理中的研究前景。该方法不依赖于阻断事件数据集的先验信息,可自主地发现数据集中的聚类簇,对于识别未知的信号种类或编码特征具有较好的研究和应用价值。
猜你喜欢
华人时刊(2022年9期)2022-09-06
网络安全与数据管理(2022年1期)2022-08-29
内燃机与动力装置(2022年1期)2022-03-21
锻压装备与制造技术(2021年5期)2021-11-13
哈尔滨工程大学学报(2021年2期)2021-03-19
科学技术创新(2021年5期)2021-03-17
华人时刊(2020年15期)2020-12-14
——编码器
演艺科技(2020年7期)2020-08-13
农业工程学报(2020年3期)2020-04-09
筑路机械与施工机械化(2017年5期)2017-08-31
