
茂金属催化剂催化丙烯聚合的β-Me 消除选择性研究
2020-07-08 07:35肖凌云马海燕
华东理工大学学报(自然科学版) 2020年3期
肖凌云, 马海燕
(华东理工大学化学与分子工程学院金属有机化学研究室,上海 200237)
丙烯是一种来源于石油的廉价且重要的工业原料,其齐聚物可以作为清洁剂、人造油和可塑剂前体,还可以作为单体参与聚合或者共聚得到高性能聚合物。聚丙烯(PP)是一种结构规整的结晶性聚合物,无味、无毒、质轻且具有热塑性,具有优良的材料性能,在工业中具有广泛的应用,产量仅次于聚乙烯和聚氯乙烯[1]。
在催化丙烯齐聚和高聚反应时,链转移反应中β-H 消除和β-巯基乙醇(β-Me)消除两种链转移方式的竞争是影响聚合产物分子量以及端基结构的重要因素[2]。有研究[3-6]表明,在利用具有大位阻取代基的茂金属催化剂催化丙烯聚合的过程中,β-Me 消除链转移反应会在一定程度上引起聚合产物分子量下降,并具有生成烯丙基端基丙烯齐聚物或低分子量聚合物的潜力,这些烯丙基端基产物能被用作均聚和共聚反应的大单体,也可以作为嵌段结构用于制备新型聚合物[6-7]。
大量烯烃聚合领域的研究表明,在丙烯聚合反应中要实现较高概率的β-Me 消除链转移反应,无论是从催化剂的空间效应还是从电子效应上进行调控,都要比β-H 消除链转移反应难以实现。Resconi 小组[3]于1992 年 报 道 了(Cp*)2MCl2/ MAO体 系(Cp*为C5Me5, M 为Zr、Hf)在50 ℃催化液化丙烯聚合得到分子量很低的产物(聚合度 ≤20),当M 分别为Zr 和Hf 时,烯丙基端基和亚乙烯基端基产物的物质的量之比分别为92∶8 和98∶2,烯丙基端基产物为β-Me 消除反应的结果,亚乙烯基端基产物则对应β-H 消除反应。随后Resconi 小组[4]于1996 年报道了络合物C2H4-(Flu)2ZrCl2经甲基铝氧烷(MAO)活化后催化丙烯聚合可得到质量分数为75%的烯丙基端基产物。1998 年Resconi 小组[5]再次报道了丙烯聚合中β-Me 消除选择性的研究,碳桥联或亚乙基桥联的双茚锆络合物rac-Me2C-(Ind)2ZrCl2和rac-C2H4-(Ind)2ZrCl2均无β-Me 消除选择性,而在其茚环上增加甲基、叔丁基和三甲基硅基后所得络合物均表现出一定的β-Me消除选择性,选择性最高为51.7%。2000 年Weng 小组[8]报道了用rac-Me2Si-(2-Me-4-Ph-Ind)2ZrCl2和rac-Me2Si-(Ind)2HfMe2两种催化剂催化丙烯聚合,得到了烯丙基端基摩尔分数为60%~80%的高等规度聚丙烯,熔点达到140~150 ℃。2002 年Janiak 小 组[9]用 (C5Me4H)2ZrCl2、 (C4Me4P)2ZrCl2和(C5Me5)2ZrCl23 种茂金属络合物催化丙烯聚合,分别得到摩尔分数为65%、61%和9%的烯丙基端基产物。2012 年,Carpentier 等[10]报道了亚甲基桥联茂-芴锆络合物[Ph(H)C]-(3,6-tBu2Flu)(3-tBu-5-Ph-C5H2)ZrCl2经MAO 活化后在80 ℃甲苯中催化丙烯聚合得到高等规立构的齐聚物,并且仅有乙烯基和亚乙烯基两种端基,其中β-Me 选择性为66%。Rieger 小组[6]于2013 年报道了一系列C1-对称亚乙基桥联茚-芴锆、铪络合物rac-C2H4-(Flu)(2-Me-5,6-cyclopenta-Ind)MCl2(M = Zr, Hf),其在较高温度条件下催化丙烯聚合均可以得到烯丙基端基产物。其中,锆催化剂体系在60 ℃时β-Me 消除选择性为64%,铪催化剂体系在70 ℃时β-Me 消除选择性达到93%。除此之外,其他具有β-Me 消除选择性的络合物催化剂鲜有报道,仅有Ln(III)[11]和Sc(III)[12]等几例络合物。而在以茂金属锆、铪络合物为主的催化剂中,可能由于高的β-Me 消除选择性难以获得,迄今为止并没有深入研究配体结构与β-Me 消除选择性之间关系的报道。
本课题组在前期研究[13]中发现,在亚乙基桥联茚-芴锆络合物的茚环3-位引入苄基后,催化剂的β-Me消除选择性大幅上升,而所得丙烯聚合物分子量有所下降。在引入苄基的基础上,在茚环5,6-位引入1,5-环戊基以及2-位引入甲基后,β-Me 选择性有所上升,但催化活性和所得产物分子量都呈下降趋势[14]。基于这一结果,本文合成了一系列亚乙基桥联茚-芴锆、铪络合物,在其茚环5,6-位引入1,5-环戊基,在部分结构茚环3-位引入苄基,考察这些结构特点对相应络合物催化丙烯聚合性能的影响,以及是否有利于获得兼具高催化活性和较高β-Me 消除选择性的络合物。
1 实验部分
1.1 主要试剂和仪器
涉及对水、氧敏感的原料、中间体或产物的实验均在氩气保护下进行,并采用标准Schlenk 操作。乙醚、四氢呋喃(THF)、甲苯、正己烷在氩气保护下加入钠丝,并以二苯甲酮为指示剂,加热回流至深紫色(乙醚、四氢呋喃、甲苯)或深蓝色(正己烷)后蒸出备用;二氯甲烷在氩气保护下加入CaH2加热回流并干燥处理后蒸出备用。上述实验原料与试剂均为分析纯,购于上海国药集团化学试剂有限公司。5,6-环戊基茚、1-苄基-5,6-环戊基茚、9-(2-溴乙基)芴、1-[9-(2,7-二叔丁基)芴基]-2-溴乙烷参照文献方法合成[15-17]。1H-NMR 和13C-NMR 采 用Bruker AVANCE-400型核磁共振仪(德国Bruker 公司)测定,以CDCl3为溶剂,四甲基硅(TMS)为内标。高分辨质谱采用GCT Premier 型高分辨质谱仪(美国 Waters 公司)测定。络合物单晶结构采用Bruker SMART 1000CCD衍射仪(德国Bruker 公司)测定。
1.2 配体的合成
1.2.1 1-(9-芴基)-2-[1-(5,6-环戊基)茚基]乙烷(L1)的合成 向100 mL 圆底Schlenk 瓶中加入1.74 g(11.14 mmol) 5,6-环戊基茚(Ind1),用50 mL 干燥乙醚溶解,在液氮-乙醇浴(–78 ℃)冷却下用注射器逐滴加入4.46 mL (11.14 mmol)nBuLi 的正己烷溶液,反应液从无色清液变成白色浑浊液,恢复室温后继续搅拌12 h。在冰浴下加入3.04 g (11.14 mmol) 9-(2-溴乙基)芴(Flu1),自然升至室温,继续搅拌12 h后,在液氮-乙醇浴(–78 ℃)冷却下加入20 mL 干燥THF,搅拌2 h,加入20 mL 饱和NH4Cl 溶液终止反应。分液,水相用20 mL CH2Cl2萃取3 次,合并有机相,加适量无水MgSO4干燥3 h,抽滤,滤液旋干,得到黄色油状粗产物。用200~300 目(75~48 μm)硅胶进行柱色谱分离,纯石油醚(沸点 60~90 ℃)为洗脱剂,得到浅黄色固体1.80 g,产率46.4%。1H-NMR (400 MHz,298 K, CDCl3, δ):7.75 (dd, J = 7.3, 3.9 Hz, 2H, Ar-H),7.51 (d, J = 6.8 Hz, 1H, Ar-H), 7.43 (d, J = 6.8 Hz, 1H,Ar-H), 7.39~7.27 (m, 4H, Ar-H), 7.18 (s, 1H, Ar-H),7.11 (s, 1H, Ar-H), 6.74 (dd, J = 5.5, 1.8 Hz, 1H, Ind C5-ring vinylic-H), 6.40 (dd, J = 5.5, 1.8 Hz, 1H, Ind C5-ring vinylic-H), 3.98 (t, J = 5.5 Hz, 1H, 9-Flu-H), 3.34~3.28(m, 1H, 1-Ind-H), 2.90 (t, J = 7.4 Hz, 4H, CH2CH2CH2),2.22~1.94 (m, 4H, CH2CH2CH2and Ind-CH2CH2-Flu),1.71 (tt, J = 12.2, 4.9 Hz, 1H, Ind-CH2CH2-Flu), 1.39~1.28(m, 1H, Ind-CH2CH2-Flu)。13C-NMR (100 MHz, 298 K,CDCl3, δ): 147.1, 147.0, 146.0, 145.4, 143.0, 142.5,141.3, 141.3, 141.0, 138.1, 131.2, 127.0, 126.92, 126.88,124.4, 119.89, 119.85, 119.1, 116.9, 49.7, 47.4, 32.8,32.7, 29.7, 26.9, 26.0。HR-MS (EI, m/z) for C27H24:Calcd., 348.187 8; Found, 348.188 0。
1.2.2 1-[9-(2, 7-二叔丁 基)芴基]-2-[1-(5, 6-环戊基)茚基]乙烷(L2)的合成 向100 mL 圆底Schlenk 瓶中加入2.20 g (14.08 mmol) 5, 6-环戊基茚(Ind1),用50 mL 干燥乙醚溶解,在液氮-乙醇浴(–78 ℃)冷却下用注射器逐滴加入5.63 mL (14.08 mmol)nBuLi 的正己烷溶液,反应液从无色清液变成白色浑浊液,恢复室温后继续搅拌12 h。在冰浴下加入4.89 g (12.67 mmol)1-[9-(2,7-二叔丁基)芴基]-2-溴乙烷(Flu2),自然升至室温,继续搅拌48 h 后,加入20 mL 饱和NH4Cl 溶液终止反应。分液,水相用20 mL CH2Cl2萃取3 次,合并有机相,加适量无水MgSO4干燥3 h,抽滤,滤液旋干,得到白色固体粗产物。用二氯甲烷和石油醚重结晶,得到白色固体5.02 g,产率77.4%。1H-NMR(400 MHz, 298 K, CDCl3, δ): 7.62 (dd, J = 8.0, 2.7 Hz,2H, Ar-H), 7.50 (d, J = 0.7 Hz, 1H, Ar-H), 7.43 (d, J =0.7 Hz, 1H, Ar-H), 7.40~7.34 (m, 2H, Ar-H), 7.19 (s,1H, Ar-H), 7.11 (s, 1H, Ar-H), 6.75 (dd, J = 5.5, 1.8 Hz,1H, Ind C5-ring vinylic-H), 6.38 (dd, J = 5.5, 1.8 Hz, 1H,Ind C5-ring vinylic-H), 3.91 (t, J = 5.3 Hz, 1H, 9-Flu-H),3.30 (m, 1H, 1-Ind-H), 2.90 (dd, J = 13.1, 6.0 Hz, 4H,CH2CH2CH2), 2.16~2.02 (m, 4H, CH2CH2CH2and Ind-CH2CH2-Flu), 1.97 (ddd, J = 18.1, 8.6, 5.1 Hz, 1H, Ind-CH2CH2-Flu), 1.70~1.59 (m, 1H, Ind-CH2CH2-Flu),1.39 (s, 9H, C(CH3)3), 1.38 (s, 9H, C(CH3)3)。13C-NMR(100 MHz,298 K, CDCl3, δ): 147.2, 147.1, 146.1, 143.0,142.6, 141.4, 141.3, 141.1, 138.2, 131.3, 127.1, 128.0,127.0, 124.5, 120.0, 119.9, 119.2, 117.0, 49.8, 47.5, 32.8,32.8, 29.8, 27.01, 26.01。HR-MS (EI, m/z) for C35H40:Calcd., 460.313 0; Found, 460.312 6。
1.2.3 1-[9-(2,7 二叔丁基)芴基]-2-[1-(3-苄基-5,6-环戊基)茚基]乙烷(L3)的合成 向100 mL 圆底Schlenk 瓶中加入2.39 g (9.70 mmol) 1-苄基-5,6-环戊基茚(Ind2),用50 mL 干燥乙醚溶解,在液氮-乙醇浴(–78 ℃)冷却下用注射器逐滴加入3.88 mL (9.70 mmol)nBuLi 的正己烷溶液。反应液从无色清液变成橙色浑浊液,恢复室温后继续搅拌12 h。在冰浴下加入3.74 g (9.70 mmol) 1-[9-(2,7-二叔丁基)芴基]-2-溴乙烷(Flu2),自然升至室温,继续搅拌12 h 后,在液氮-乙醇浴(–78 ℃)冷却下加入20 mL 干燥THF,搅拌2 h,加入20 mL 水终止反应。分液,水相用20 mL CH2Cl2萃取3 次,合并有机相,加适量无水MgSO4干燥3 h。抽滤,滤液旋干,得到黄色油状粗产物。用200~300 目(75~48 μm)硅胶进行柱色谱分离,乙酸乙酯和石油醚混合溶剂为洗脱剂,得到浅黄色固体3.05 g,产率57.0%。固体为3 种异构体(L3a~L3c)的混合物,以L3a 为主。L3a:1H-NMR (400 MHz, 298 K,CDCl3, δ): 7.62 (dd, J = 8.0, 3.3 Hz, 2H, Ar-H), 7.50 (d,J = 0.7, 1H, Ar-H), 7.43 (d, J = 0.7, 1H, Ar-H),7.40~7.34 (m, 3H, Ar-H), 7.30 (m, 1H, Ar-H), 7.28 (m,3H, Ar-H), 7.11 (s, 1H, Ar-H), 7.06 (s, 1H, Ar-H), 6.03(d, J = 1.4 Hz, 1H, Ind C5-ring vinylic-H), 3.92 (t, 1H, 9-Flu-H), 3.86 (s, 2H, PhCH2), 3.29 (m, 1H, 1-Ind-H),2.95~2.80 (m, 4H, CH2CH2CH2), 2.18~1.93 (m, 5H,CH2CH2CH2and Ind-CH2CH2-Flu), 1.66 (m, 1H, Ind-CH2CH2-Flu), 1.39 (s, 9H, C(CH3)3), 1.38 (s, 9H,C(CH3)3)。13C-NMR (100 MHz, 298 K, CDCl3, δ):149.72, 149.67, 147.3, 147.2, 147.0, 143.4, 142.3, 141.2,139.6, 138.83, 138.77, 134.6, 129.0, 128.5, 126.1,123.99, 123.98, 121.2, 121.1, 119.13, 119.09, 119.0,115.5, 48.4, 47.5, 34.99, 34.97, 34.6, 32.8, 31.8, 29.6,26.9, 26.0。HR-MS (EI, m/z) for C42H46:Calcd., 550.360 0;Found, 550.359 9。
1.3 锆、铪络合物的合成
1.3.1 {C2H4(5,6-cyclopenta-Ind)(Flu)}ZrCl2(C1)的 合成 将1.09 g (3.13 mmol)配体L1 加入100 mL 圆底Schlenk 瓶中,加入50 mL 干燥乙醚溶解,在液氮-乙醇浴(–78 ℃)冷却下通过注射器逐滴加入2.50 mL(6.16 mmol)nBuLi 的正己烷溶液,自然升至室温后继续搅拌48 h。在冰浴冷却下加入0.729 g (3.13 mmol)ZrCl4,自然升至室温,继续搅拌。48 h 后停止反应,真空除去乙醚,加入35 mL 干燥二氯甲烷,搅拌2 h后离心,得到红色溶液。真空下浓缩至干后用干燥甲苯溶解,加入正己烷调至微混浊,放入–20 ℃冰箱中重结晶。分离得到红色固体0.32 g,产率为20.1%。1H-NMR (400 MHz, CDCl3, 298 K,δ):7.95 (d,J = 8.4 Hz, 1H, Ar-H), 7.86 (d, J = 8.4 Hz, 1H, Ar-H),7.80 (d, J = 8.5 Hz, 1H, Ar-H), 7.62 (d, J = 8.4 Hz, 1H,Ar-H), 7.59~7.54 (m, 1H, Ar-H), 7.50 (s, 1H, Ar-H),7.42~7.31 (m, 2H, Ar-H), 7.20 (m, 1H, Ar-H), 7.15 (s,1H, Ar-H), 6.20 (d, J = 3.4 Hz, 1H, Ind C5-ring vinylic-H), 6.12 (d, J = 3.4 Hz, 1H, Ind C5-ring vinylic-H),4.41~4.30 (m, 1H, Ind-CH2CH2-Flu), 4.13~3.97 (m, 2H,Ind-CH2CH2-Flu), 3.95~3.84 (m, 1H, Ind-CH2CH2-Flu),2.95~2.76 (m, 4H, CH2CH2CH2)), 2.06~1.91 (m, 2H,CH2CH2CH2)。13C-NMR (100 MHz, 298 K, CDCl3, δ):145.8, 128.6, 125.5, 125.2, 124.9, 124.7, 124.0, 122.9,122.7, 121.2, 119.4, 115.8, 112.6, 107.4, 32.6, 32.3, 29.7,26.4。HR-MS (EI, m/z) for C27H22ZrCl2: Calcd. 508.011 6; Found, 508.012 8。
1.3.2 {C2H4(5,6-cyclopenta-Ind)(2,7-tBu2-Flu)} ZrCl2(C2)的合成 将1.05 g (2.28 mmol) 配体L2 加入100 mL圆底Schlenk 瓶中,加入50 mL 干燥乙醚溶解,在液氮-乙醇浴(–78 ℃)冷却下通过注射器逐滴加入1.82 mL(4.56 mmol)nBuLi 的正己烷溶液,自然升至室温后继续搅拌48 h。在冰浴冷却下加入0.531 g (2.28 mmol)ZrCl4,自然升至室温,继续搅拌。4 d 后停止反应,真空除去乙醚,加入35 mL 干燥二氯甲烷,搅拌2 h 后离心,得到红色溶液。抽干后用干燥二氯甲烷溶解,加入正己烷调至微混浊,放入–20 ℃冰箱中重结晶。最后分离得到红色固体0.41 g,产率 29.0 %。1H-NMR (400 MHz, CDCl3, 298 K, δ):7.81 (dd, J = 8.4,1.3 Hz, 1H, Ar-H), 7.72 (dd, J = 8.8, 0.6 Hz, 1H, Ar-H),7.67~7.62 (m, 2H, Ar-H), 7.53 (s, 2H, Ar-H), 7.39 (dd,J = 8.8, 1.6 Hz, 1H, Ar-H), 7.15 (s, 1H, Ar-H), 6.19 (d, J =3.3 Hz, 1H, Ind C5-ring vinylic-H), 5.97 (d, J = 3.3 Hz,1H, Ind C5-ring vinylic-H), 4.37 (ddd, J = 14.3, 11.9, 7.6 Hz, 1H, Ind-CH2CH2-Flu), 4.12~3.96 (m, 2H, Ind-CH2CH2-Flu), 3.89 (ddd, J = 21.2, 11.8, 7.6 Hz, 1H, Ind-CH2CH2-Flu), 2.95~2.75 (m, 4H, CH2CH2CH2), 1.97 (p,J = 7.2 Hz, 2H, CH2CH2CH2), 1.43 (s, 9H, C(CH3)3),1.31 (s, 9H, C(CH3)3)。13C-NMR (100 MHz, 298 K,CDCl3, δ): 151.4, 151.2, 145.5, 144.8, 125.0, 124.2,123.1, 121.7, 119.3, 115.9, 111.7, 102.7, 35.4, 35.3, 32.8,32.3, 31.3, 31.1, 29.9, 29.8, 26.5。HR-MS (EI, m/z) for C35H38Cl2Zr: Calcd., 620.139 7; Found, 620.140 1。
1.3.3 {C2H4(3-Bn-5,6-cyclopenta-Ind)(2,7-tBu2-Flu)}ZrCl2(C3)的合成 将1.01 g (1.83 mmol) 配体L3 加入100 mL 圆底Schlenk 瓶中,加入50 mL 干燥乙醚溶解,在液氮-乙醇浴(–78 ℃)冷却下通过注射器逐滴加入1.36 mL (3.40 mmol)nBuLi 的正己烷溶液,自然升至室温后继续搅拌36 h。在冰浴冷却下加入0.396 g (1.70 mmol) ZrCl4,自然升至室温,继续搅拌。48 h 后停止反应,真空除去乙醚,加入45 mL 干燥二氯甲烷,搅拌2 h 后离心,得到红色溶液。抽干后用干燥二氯甲烷溶解,加入正己烷调至微混浊,放入–20 ℃冰箱中重结晶。最后分离得到橙红色固体0.25 g,产 率42.4%。1H-NMR (400 MHz, CDCl3, 298 K, δ):7.84 (d, J = 8.3 Hz, 1H, Ar-H), 7.76 (d, J = 8.9 Hz,1H, Ar-H), 7.65~7.60 (m, 2H, Ar-H), 7.53 (s, 1H, Ar-H),7.41 (dd, J = 8.8, 1.6 Hz, 1H, Ar-H), 7.39 (s, 1H, Ar-H),7.19~7.09 (m, 3H, Ar-H), 7.07 (s, 1H, Ar-H), 7.02~6.98(m, 2H, Ar-H), 5.79 (s, 1H, Ind C5-ring vinylic-H), 4.20(ddt, J = 22.1, 14.7, 7.5 Hz, 2H, Ind-CH2CH2-Flu), 4.07~3.92(m, 2H, CH2Ph and Ind-CH2CH2-Flu), 3.84~3.67 (m, 2H,CH2Ph and Ind-CH2CH2-Flu), 2.92~2.71 (m, 4H,CH2CH2CH2), 2.01~1.87 (m, 2H, CH2CH2CH2), 1.42 (s,9H, C(CH3)3), 1.34 (s, 9H, C(CH3)3)。13C-NMR (100 MHz, 298 K, CDCl3, δ): 151.3, 145.3, 145.0, 141.2,128.5, 125.8, 125.7, 124.8, 124.5, 124.3, 124.1, 120.3,117.2, 115.5, 112.0, 102.9, 35.4, 35.3, 34.5, 32.7, 32.4,31.29, 31.1, 29.7, 29.2, 26.6。 HR-MS (EI, m/z) for C42H44ZrCl2:Calcd., 710.187 0; Found, 710.185 9。
1.3.4 {C2H4(3-Bn-5,6-cyclopenta-Ind) (2,7-tBu2-Flu)}HfCl2(C4)的 合 成 将1.03 g (1.87 mmol) 配体L3加入100 mL 圆底Schlenk 瓶中,加入50 mL 干燥乙醚溶解,在液氮-乙醇浴(–78 ℃)冷却下通过注射器逐滴加入1.39 mL (3.49 mmol)nBuLi 的正己烷溶液,自然升至室温后继续搅拌48 h。在冰浴冷却下加入0.558 g (1.74 mmol) HfCl4,自 然 升 至室温,继续搅拌。48 h 后停止反应,真空除去乙醚,加入45 mL 干燥二氯甲烷,搅拌2 h 后离心,得到红色溶液。抽干后用干燥甲苯溶解,加入正己烷调至微混浊,再放入–20 ℃冰箱中重结晶。最后分离得到橙黄色固体0.248 g,产率16.6%。1H-NMR (400 MHz, CDCl3, 298 K,δ):7.84 (d, J = 8.9 Hz, 1H, Ar-H), 7.76 (d, J = 8.9 Hz,1H, Ar-H), 7.58 (dd, J = 8.9, 1.6 Hz, 1H, Ar-H), 7.55 (s,1H, Ar-H), 7.52 (s, 1H, Ar-H), 7.38 (dd, J = 8.8, 1.6 Hz,2H, Ar-H), 7.20~7.09 (m, 3H, Ar-H), 7.06 (s, 1H, Ar-H),7.02~6.97 (m, 2H, Ar-H), 5.74 (s, 1H, Ind C5-ring vinylic-H), 4.35~4.26 (m, 1H, Ind-CH2CH2-Flu), 4.23~4.14(m, 1H, Ind-CH2CH2-Flu), 4.09 (dd, J = 14.2, 7.3 Hz,1H, Ind-CH2CH2-Flu), 4.03 (d, J = 16.0 Hz, 1H, CH2Ph),3.90~3.82 (m, 1H, Ind-CH2CH2-Flu), 3.79 (d, J = 16.1 Hz,1H, CH2Ph), 2.95~2.75 (m, 4H, CH2CH2CH2), 2.00~1.87(m, 2H, CH2CH2CH2), 1.41 (s, 9H, C(CH3)3), 1.33 (s,9H, C(CH3)3)。13C-NMR (100 MHz, 298 K, CDCl3,δ):151.0, 150.9, 145.1, 144.7, 141.4, 128.5, 128.4, 126.8,126.7, 126.1, 125.0, 124.51, 124.5, 124.1, 124.0, 120.2,119.4, 117.9, 117.5, 117.1, 116.8, 115.8, 115.2, 110.6,98.9, 35.4, 35.3, 34.4, 32.59, 32.4, 31.1, 31.2 29.1, 28.5,26.6。HR-MS (EI, m/z) for C42H44HfCl2:Calcd., 798.228 6;Found, 798.227 7。
1.4 络合物C2 晶体结构的测定
络合物C2 的单晶是从其饱和的二氯甲烷/正己烷混合溶液中于–20 ℃培养得到,呈红色棱柱形。X 射线单晶衍射测试采用Bruker SMART APEX II 衍射仪完成,石墨单色化Mo-Kα 射线(λ= 0.710 73×10−10m)。相 关 计 算 采 用SHELXS-97 和SHELXL-97 程序完成,分子空间结构图使用Ortep3v2 程序绘制。
1.5 丙烯聚合实验
以C2 催化丙烯聚合为例,在铝、锆物质的量之比为4 000,丙烯单体压力为0.4 MPa 时使用50 mL安培瓶,通过标准Schlenk 操作方法,称取12.5 mg 络合物C2,用干燥注射器加入16.1 mL 干燥甲苯,配制成浓度为1.25 × 10–3mol/L 的催化剂溶液。
将搅拌子放入50 mL 玻璃内衬里,将玻璃内衬置于高压釜内并将釜体密封,保持搅拌子均匀搅拌,调节油浴锅温度至约180 ℃抽烤高压釜,保持30 min后置换丙烯气体,重复3 次。抽烤结束后置换成丙烯气体,油浴锅调温至100 ℃。达到设定温度后在丙烯保护下打开加料口,依次加入19.0 mL 甲苯和5.0 mL浓度为1.03 mol/L MAO 的甲苯溶液,关闭加料口,恒温20 min 后加入1 mL 催化剂溶液,再关闭加料口,调整丙烯压力到0.40 MPa,并开始计时。30 min 后停止搅拌和加热,关闭高压釜进气口,并用冰盐浴冷却高压釜。释放釜内压力,快速取出玻璃内衬,冰盐浴冷却下边搅拌边加入200 μL 内标正庚烷,然后再缓慢加入2.0 mL 甲醇,充分搅拌后取少量液体经过滤后封装于密闭玻璃管内,用于低沸点齐聚物组分的分析。在剩下的反应液中缓慢滴加盐酸、乙醇(体积比3∶97)混合溶液终止反应,再充分搅拌1 h 以上。分液,将甲苯相与乙醇相分开,甲苯相水洗3 次,洗液与乙醇相合并后用甲苯再次萃取,最后将萃取液与甲苯相合并,加适量无水硫酸镁干燥后过滤。旋蒸除去溶剂得到聚合产物,进一步在60 ℃下真空干燥12 h 后称重。根据聚合产物的氢核磁谱图计算出聚合物的分子量以及烯丙基端基的质量分数。
2 结果与讨论
2.1 络合物的合成和表征
根据本组之前沿用的方法[14],将取代茚上的活泼氢用nBuLi 攫取后得到相应锂盐,然后与Flu1 或Flu2 发生亲核取代反应得到目标亚乙基桥联取代茚-芴配体L1~L3,其结构如图1(a)所示。
在L3 合成过程中,由于茚环3-位具有取代基,反应所得产物是L3a、L3b 和L3c 3 种异构体的混合物(图1(b))。其中L3a 和L3b 在nBuLi 的作用下均可生成配体的双锂盐,所以并不影响目标络合物的合成;而L3c 的茚环上没有活泼氢,碳原子连接顺序也与目标化合物不同,故视作配体合成过程中的副产物。
进一步研究发现,在配体合成过程中溶剂的选择对反应的活性和选择性影响很大。当使用乙醚为溶剂时,副产物L3c 的生成会得到较好的抑制,但反应活性很低;而使用THF 为溶剂,反应活性得到明显的提高,但副产物L3c 的含量也明显增加。综合考虑以上因素并结合本课题组其他研究者的经验[18],采用分阶段使用乙醚/THF 混合溶剂的方法,即先在乙醚中反应24 h,然后在低温条件下缓慢加入少量THF 继续反应直至反应结束。在实际操作中,由于L3c 的性质与L3a、L3b 接近,很难将其除去。
配体与nBuLi 反应生成双锂盐后与ZrCl4或HfCl4发生转金属化反应得到目标锆络合物C1~C3以及铪络合物C4,通过重结晶分离纯化,其结构如图1(c)所示。络合物C2 在重结晶过程中得到了单晶,其分子结构如图2 所示,部分键长和键角数据见表1。从单晶数据可以看出,金属中心与茚环和芴环均为η5配位,但由于茚环5,6-位引入1,5-环戊基后,其与芴环同侧叔丁基存在一定排斥作用,使得金属中心到茚环、芴环五元环部分各碳原子的键长相差显著,茚环-芴环平面更为张开,二面角角度变大[14]。
在利用该类配体合成络合物的过程中,茚基和芴基五元环均失去一个氢质子,因此在络合物的1HNMR 谱图中不会出现相应氢的信号;此外,由于络合物中的茚环和芴环对亚乙基桥的氢具有去屏蔽作用,其信号相对于自由配体中相应的氢信号明显向低场移动,如配体L2 及相应络合物C2 的核磁氢谱所示(图3)。另外从图3 中可以看出,L2 中芴环上两个叔丁基的两组峰并未完全分开,而C2 中两个叔丁基的氢信号呈两组非常明显的单峰,并且C2 中各芳基的氢信号也表现出更为明显的位移差别。这是由于配体与中心金属成键后,茚环和芴环无法围绕亚乙基桥自由旋转,说明络合物具有C1-对称性。本文中络合物C1、C3、C4 的1H-NMR 谱图也表现出类似的特点。
2.2 催化丙烯聚合研究
以络合物C1~C4 为主催化剂,MAO 为助催化剂,催化丙烯聚合的结果见表2。聚合产物的1HNMR 分析表明产物中仅含有烯丙基端基和亚乙烯基端基这两种不饱和端基(如图4 所示),这两种不饱和端基分别由丙烯单体经1,2-插入后发生β-Me 消除反应和β-H 消除反应形成(如图5 所示)[19]。因此,讨论烯丙基端基的含量即等同于讨论β-Me 消除选择性。
从表2 络合物C1~C4 催化丙烯聚合的结果可以看出,络合物配体结构以及中心金属的改变对相应络合物在不同条件下催化丙烯聚合的性能影响很大,其催化活性、所得聚合产物分子量以及烯丙基端基含量即β-Me 消除选择性等都有较为显著的变化。

图1 配体L1~L3 的合成路线(a);配体合成过程中形成的异构体(b);络合物C1~C4 的合成路线(c)Fig. 1 Synthetic route of ligands L1~L3(a); Isomers formed in the synthesis of ligands(b); Synthetic route of complexes C1~C4

图2 络合物C2 的分子结构Fig. 2 Molecular structure of complex C2
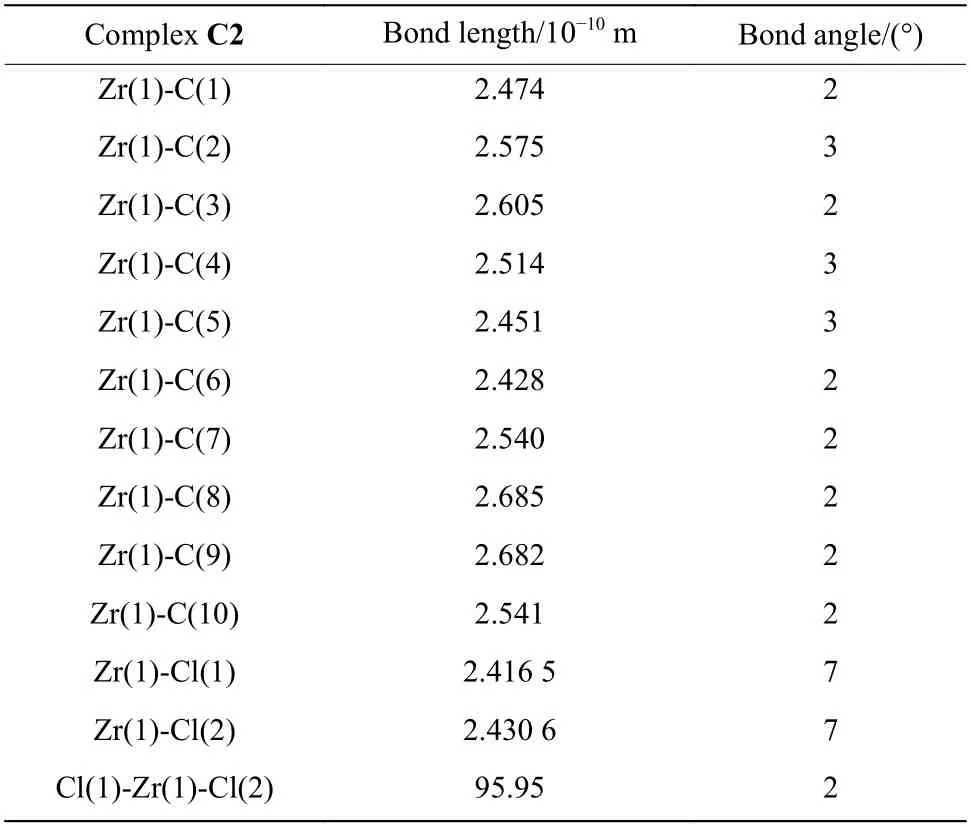
表1 络合物C2 的部分键长和键角Table 1 Selected bond lengths and angles for complex C2
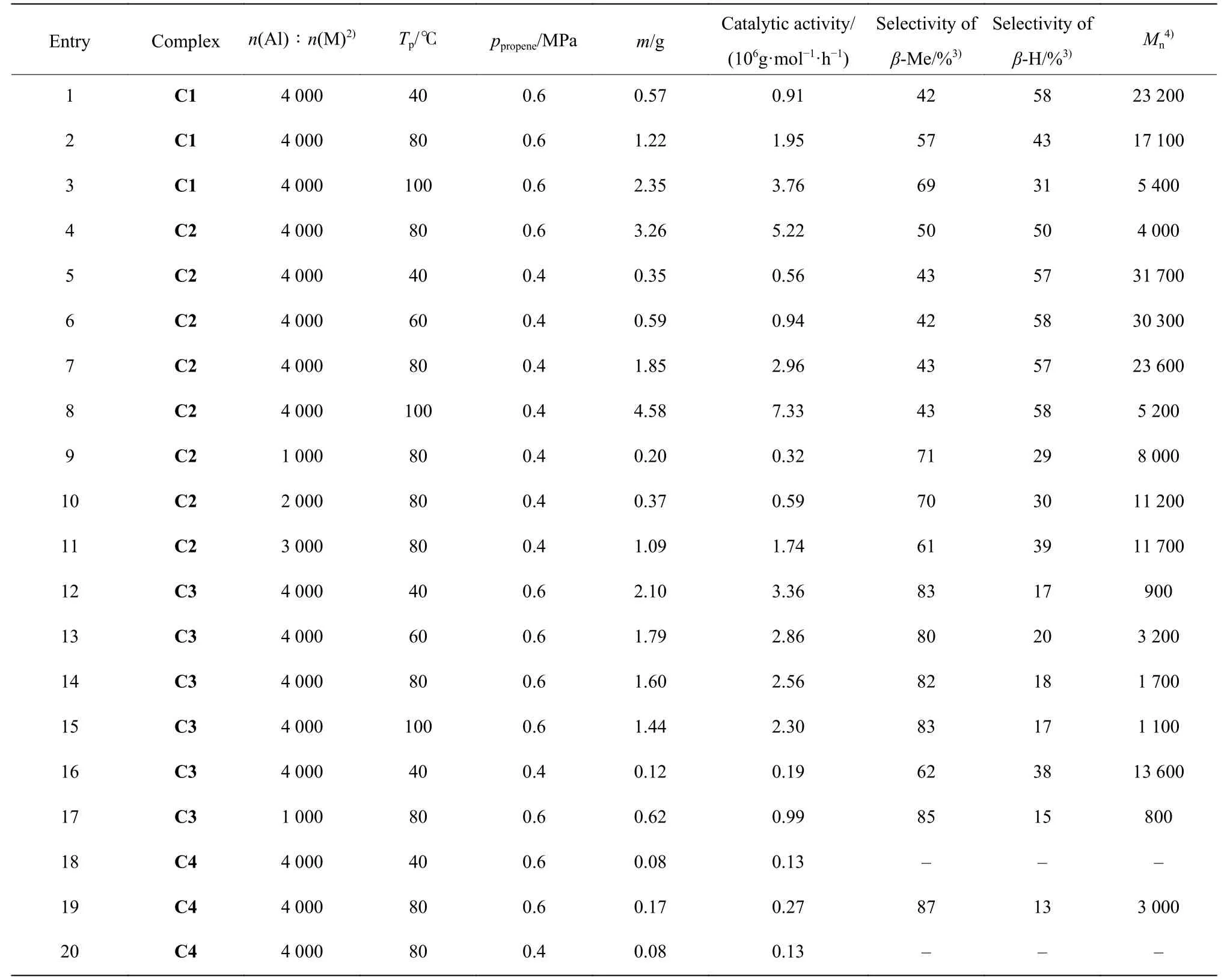
表2 络合物C1~C4 催化丙烯聚合结果1)Table 2 Propene polymerization catalyzed by complexes C1~C41)
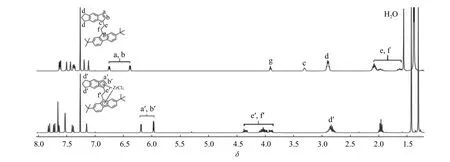
图3 配体L2 和络合物C2 的1H-NMR 谱图(CDCl3, 400 MHz)Fig. 3 1H-NMR spectra of ligand L2 and complex C2 (CDCl3, 400 MHz)
锆络合物C1 中,除茚环5,6-位环戊基取代外,茚环和芴环上没有其他取代基,该络合物在40~100 ℃催化丙烯聚合时(Entry 1、2、3),表现出较高的催化活性,且随温度上升,催化活性增加,在100 ℃时催化活性达到最高,为3.76 × 106g/(mol·h)。在络合物芴环2,7-位引入叔丁基后,锆络合物C2 的催化活性显著增加,是络合物C1 的两倍以上(Entry 2、4)。与络合物C1 类似,络合物C2 的催化活性随聚合温度升高而增加,在100 ℃时达到最高值(Entry 5~8)。芴环上2,7-位叔丁基的引入在一定程度上增加了络合物金属中心的空间位阻,丙烯分子对金属中心1,2-位插入时位阻增大,导致活性降低;但另一方面,2,7-位叔丁基的供电子能力使得金属中心的电子密度增加,从而使活性中心的稳定性增强,有利于催化活性的提高[14]。基于络合物C2 在40~60 ℃温度条件下催化活性偏低,而在较高温度时(80~100 ℃)催化活性增加显著,由此我们认为芴环2,7-位叔丁基在较低温度时对络合物催化性能的影响以空间效应为主,而在较高温度(80~100 ℃)时以电子效应为主。

图4 丙烯聚合产物的1H-NMR 谱图(CDCl3, 400 MHz)Fig. 4 1H-NMR spectrum of propylene polymerization product (CDCl3, 400 MHz)

图5 亚乙基桥联取代茚-芴体系催化丙烯齐聚/聚合形成的主要端基结构Fig. 5 Structures of dorminant terminal groups formed in propylene oligomerization/polymerization catalyzed by ethylene-bridged substituted indenyl-fluorenyl metal complexes
分析络合物C1 及芴环2,7-位引入叔丁基后的络合物C2 催化丙烯聚合所得产物的端基成分发现,芴环2,7-位引入叔丁基后,催化剂的β-Me 消除选择性的变化趋势发生了很大的变化。芴环不含叔丁基取代的络合物C1 其β-Me 消除选择性随聚合温度升高而逐渐增加,在40~100 ℃范围内为42%~69%。而络合物C2 的β-Me 消除选择性受温度影响很小,在40~100 ℃基本维持不变(Entry 5~8),且低于同等条件下C1 的选择性(Entry 2 对比Entry 3)。因此可以认为在芴环2,7-位引入叔丁基不利于在聚合过程中发生β-Me 消除链转移反应。
对比Entry 4~8 和Entry 12~16 发现,进一步在茚环3-位引入苄基对相应锆络合物C3 的催化活性和催化所得产物的分子量均有显著影响。在MAO 活化下,茚环3-位不含苄基的络合物C2 催化活性随聚合温度升高而增加;且其催化丙烯聚合所得产物聚丙烯均为半透明膜状或块状,在40~100 ℃范围内,聚合物分子量随温度升高而降低,在40 ℃时所得到的聚合产物分子量最高,Mn达到31 700。而络合物C3 的催化活性随温度升高反而有所降低,总体上其催化活性低于络合物C2。此外,络合物C3 催化丙烯聚合所得聚合物分子量与络合物C2 相比大幅下降,产物为半透明黏液或膜状聚丙烯,在60 ℃时分子量达到最高(Mn= 3 200)。络合物C2 和C3 在烯丙基端基选择性上的差异更为明显。络合物C2 的β-Me 消除选择性在40~100 ℃范围内基本无变化;络合物C3 的消除选择性受温度影响也较小,在40~100 ℃时β-Me 消除选择性达到80%~83%,但显著高于络合物C2。
文献[11, 20-25]报道,在茂金属络合物的茂环或茚环3-位引入取代基后,在聚合条件下取代基上的氢有可能与金属中心发生分子内C—H 键活化而形成M—C 键,使活性中心失去催化烯烃聚合的活性。从表2数据可知络合物C3 构成的催化体系并不存在此种情况。我们认为茚环3-位引入苄基后,苄基因其空间位阻[26-28]或潜在的π-配位能力[17,29-30]会增加丙烯对金属中心配位插入的难度,对丙烯催化不利;另一方面,苄基潜在的π-配位能力可能有利于削弱活性物种阳离子与助催化剂阴离子之间的相互作用,从而提高催化剂的活性[31-33]。由此,C3 催化活性的改变可能是多种因素综合影响所致,但以位阻效应为主。
茂金属催化剂催化烯烃聚合反应中,链增长和链终止的相对速率是产物分子量的决定因素。3-位苄基的空间位阻对链增长速率的影响不仅降低了C3 的催化活性,也会导致C3 催化丙烯所得聚合物的分子量有所下降。然而,络合物C3 在40~100 ℃范围内催化丙烯聚合所得产物分子量显著低于络合物C2 所得产物,这一现象说明茚环3-位苄基的引入使得聚合过程中的总体链终止速率大为提高。同时,络合物C3 催化丙烯聚合过程中具有明显较高的β-Me 消除选择性,因此认为C3 催化丙烯聚合过程中链终止反应速率较快的特点可能与β-Me 消除反应有关。β-Me 消除反应对分子量的抑制作用在之前的研究中也有报道[3-6]。
与本组已报道[13]的类似结构络合物相比,络合物C3 茚环2-位没有甲基但仍保留了3-位苄基取代,该结构因素使得C3 在保持了较高β-Me 消除选择性的同时,所得聚合物分子量有较显著的下降。可见茚环3-位苄基取代是实现β-Me 高消除选择性的关键结构因素,而茚环2-位取代基则在一定程度上抑制链转移反应。
对比表2 中Entry 14 和Entry 19 可知,在MAO活化下,锆络合物C3 表现出的催化活性远高于相同配体的铪络合物C4 体系,但铪络合物C4 的β-Me 消除选择性比锆络合物C3 高约5%。
分析表2 中络合物C2 的聚合数据(Entry 7、9、10、11)发现,MAO 用量对催化剂活性、聚合物分子量和端基选择性影响显著。在n(Al)∶n(Zr)从1 000升高至4 000 的过程中,其活性由0.32 × 106g/(mol·h)升高至2.96× 106g/(mol·h),聚合物分子量由8 000 增加到23 600;而β-Me 消除选择性的变化趋势却是相反的,从71%降低至43%。同样的趋势出现在络合物C3 的催化过程中,当n(Al)∶n(Zr)由1 000 升高至4 000(Entry 17、14)时,活性由0.99 × 106g/(mol·h)升高至2.56 × 106g/(mol·h),分子量由800 增加至1 700,β-Me 消除选择性略有下降,由85%降至82%。
MAO 用量增加导致催化剂活性增大,这种现象在MAO 金属催化剂催化烯烃聚合的反应中普遍存在。n(Al)∶n(Zr)越高,阳离子活性物种与助催化剂阴离子之间的作用越弱,越有利于烯烃的配位插入;但随着n(Al)∶n(Zr)增大到一定程度,活性物种阳离子趋于游离,其活性受n(Al)∶n(Zr)的影响也变小。不过在本文采用的n(Al)∶n(Zr)为1 000~4 000 范围内,并未出现催化剂活性随n(Al)∶n(Zr)增加趋于平缓的趋势。
聚合产物分子量随n(Al)∶n(Zr)增加而增大,说明在聚合过程中基本不存在活性聚合物链向铝中心的转移,该链转移反应将导致生成饱和端基聚合物链。在我们之前的研究中,n(Al)∶n(Zr)变化对催化剂β-Me消除选择性的影响很小,而络合物C2 和C3 所表现出来的催化特性与本课题组之前报道的结果有所不同[34-35],在n(Al)∶n(Zr)较高时,β-Me 消除选择性显著降低,这可能是不同n(Al)∶n(Zr)条件下活性物种阴阳离子对的空间和电子环境的差异引起的。
分别对比表2 中Entry 4、7 和Entry 12、16 的结果发现,络合物C2 和C3 在固定温度和n(Al)∶n(Zr)不变的情况下,在丙烯单体压力从0.4 MPa 上升至0.6 MPa 的过程中,络合物的催化活性都大幅增加,聚合物的分子量大幅下降,同时β-Me 消除选择性有所提高。增大丙烯压力实际上是增加了聚合溶液体系中丙烯单体的浓度,这有利于丙烯单体对金属中心的配位插入,但其聚合物分子量显著降低,说明聚合过程中的链终止反应与链增长反应相比拥有更高的反应级数;β-Me 消除选择性的增加说明在链终止反应中,β-Me 消除反应比β-H 消除反应的级数高。有理由认为C2 或C3 催化丙烯聚合时,其β-Me 消除链转移反应可能是一个丙烯单体参与的双分子过程[2]。
综上所述,认为茚环3-位苄基取代且芴环不具有叔丁基取代的锆络合物C5 将可能兼具较好的催化活性和较高的β-Me 消除选择性。
3 结论与展望
(1)在考察的系列亚乙烯基桥联取代茚-芴锆、铪络合物中,除了以铪为金属中心的络合物C4 以外,均具有较高的催化丙烯聚合活性,而C4 具有最高的β-Me 消除选择性(87%)。
(2)络合物C1、C2 和C3 的聚合结果表明,茚环3-位引入苄基和芴环2,7-位去除叔丁基均有利于提高该系列以亚乙基桥联茚-芴结构为骨架的络合物的β-Me 消除选择性。
(3)为了更进一步增加β-Me 消除选择性,设计了茚环含苄基、芴环无叔丁基的锆络合物C5,但由于其较敏感,且在合成过程中难以与其他副产物完全分离,故未完成进一步的催化实验,后续可以尝试合成并进一步研究。
(4)如果存在较好的分离方法,可以将聚合产物中烯丙基端基聚合物与其他端基产物分离,利用分离出的烯丙基端基丙烯齐聚物/聚合物作为大分子单体与其他单体共聚,不失为一种发现材料的方法。
猜你喜欢
无机化学学报(2022年9期)2022-09-16
大众文艺(2022年16期)2022-09-07
农业工程学报(2022年5期)2022-06-22
化工时刊(2022年1期)2022-05-25
太原理工大学学报(2022年2期)2022-03-21
中国民族美术(2021年4期)2021-07-14
农药科学与管理(2019年5期)2019-08-13
画刊(2018年2期)2018-03-06
科技创新与应用(2017年20期)2017-07-15
化学教学(2016年10期)2016-11-25
