
室内饲养与野外采集的松墨天牛幼虫microRNA表达谱比较分析
2020-07-07 07:16杨炳琰赵莉蔺
生物安全学报 2020年2期
杨炳琰, 赵莉蔺*
1中国科学院动物研究所,农业虫害鼠害综合治理研究国家重点实验室,北京100101;2中国科学院生物互作卓越创新中心,中国科学院大学,北京100049
松墨天牛Monochamus alternatusHope又名松褐天牛,属鞘翅目Coleoptera天牛科Cerambycidae沟胫天牛亚科Lamiinae墨天牛属Monochamus(宋士涵和崔锡明,1991)。寄主有马尾松Pinus massonianaLamb.、黑松P.thunbergiiParl.、湿地松P.taedaL.、云南松P.yun-nanensisFranch.等,也会危害雪松Cedrus deodara(Roxb.)G.Don、冷杉Abies fabri(Mast.)Craib和落叶松Larix gmelinii(Rupr.)Kuzen(吕传海等,2000),是林业上重要的蛀干害虫。松墨天牛幼虫时期钻蛀树干和大枝条,取食韧皮部与木质部(Wuet al.,2016),虫口密度较大时可导致松树死亡。此外,松墨天牛还是国际公认的重要检疫性有害生物松材线虫Bursaphelenchus xylophilus(Steiner et Bührer)Nickel的主要传播媒介,威胁全球森林生态系统(徐华潮等,2012)。自20世纪80年代初松材线虫侵入我国境内,松墨天牛协助松材线虫扩散,最终暴发成灾,使松材线虫病成为我国最危险的林业病害(骆有庆,2001)。
松墨天牛的老熟幼虫化蛹时,会吸引大量松材线虫向其聚集,聚集在蛹室的4龄线虫被松墨天牛携带,随着天牛补充营养或产卵侵染到健康的松树中(杨宝君等,2003; Zhaoet al.,2014)。 此外,调节线虫和松墨天牛发育的信息素ascarosides在野外松墨天牛老熟幼虫体内起重要作用(Zhaoet al.,2016)。因此,对松墨天牛幼虫期的研究将有助于研究松材线虫和松墨天牛的种间互作及ascarosides的合成机理,并有助于松材线虫病的早期监测及传播阻断(陈井荣和彭小忠,2014)。然而,室内饲养与野外捕获的松墨天牛在发育速率、产卵、寿命、体型大小等方面有较明显的差异(吴桂康等,2019;徐金华等,2009)。发生这一现象的表观遗传响应机制尚不明确。因此,本实验通过研究microRNA的表达谱,以期为室内饲养和野外松墨天牛幼虫之间的代谢差异以及对ascarosides合成差异的研究提供新的思路与方法。
MicroRNAs(miRNA)由约22个核苷酸(nt)组成,是一类内源性非编码的单链小分子RNA,在动植物中广泛表达,且结构保守(Ambros,2004;Bartel,2004; Carrington&Ambros,2003; Lauet al.,2001)。研究发现,它们在多种生物过程中发挥关键作用,包括细胞分化、凋亡、增殖和脂肪储存,并且可能在昆虫对病毒抗性的产生中起作用(陈研等,2019;鲁莎等,2019; 史宏利等,2019;Chenet al.,2010; Xuet al.,2003)。 因此,松墨天牛 miRNA的测序鉴定,对于室内饲养与野外天牛发育与代谢的表观遗传调控研究将有很大帮助。
本实验通过构建4个miRNA文库,2个南京采集的野外松墨天牛幼虫组织样品和2个实验室饲养的松墨天牛幼虫组织样品,使用illuminaHiseq高通量测序,通过目标序列分类注释,获得样品中包含的各组分及表达量信息。对未注释的小RNA片段进行新miRNA的预测;对保守miRNA和新miRNA进行差异分析、靶基因预测和靶基因的GO功能注释以及KEGG pathway注释,旨在为松墨天牛幼虫发育与代谢的表观调控机制研究提供一定的参考依据。
1 材料与方法
1.1 实验材料
野外松墨天牛是2016年10月于南京(江苏省林科院提供)林间捕获的5龄老熟幼虫(NJ)。实验室饲养的松墨天牛是在南京采回来的松墨天牛进行实验室(温度25℃,湿度35%,无光照)内饲养10代的5龄老熟幼虫(LKY)。将2种天牛分别在无菌条件下解剖,获得表皮(epidermis,Ep)和中肠(mgmidgut, Mg)的样本。
1.2 文库构建及测序
使用 RNeasy Micro Kit试剂盒(Qiagen,German)提取总RNA,并使用DNaseI消化DNA后,用Urea-PAGE纯化总RNA中的sRNA,在纯化后的sRNA的3′末端和5′末端分别连接特异性的接头,再以已连接接头的sRNA为模板,逆转录合成cDNA,再通过PCR扩增,从而完成整个文库制备工作。构建文库,使用Agilent 2100 Bioanalyzer和ABI StepOnePlus Real-Time PCR System对其进行质量和产量的检测,因为松墨天牛的基因组数据尚未完全公布,所以本文参考光肩星天牛Anoplophora glabripennisMotsch的基因组数据(https:∥www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?id=217634)。最后用HiSeq 2000(illumina®,San Diego,CA,USA)平台对cDNA文库测序。一系列检测及测序由深圳华大基因科技有限公司完成。
1.3 数据过滤及分类注释
去掉接头序列、污染、低拷贝、低质量序列,得到可信的clean reads,统计小RNA(sRNA)的序列种类(unique)及序列数量(total),并对sRNA做序列的长度和数量统计。将处理后的clean reads与各类RNA进行Genbank(http:∥ftp.ncbi.nlm.nih.gov/genbank/)和Rfam11.0(http:∥rfam.janelia.org/)比对和注释,由于某些小RNA可能会比对上多个不同RNA的注释结果,按照rRNAetc>known miRNA>piRNA>repeat>exon>intron的优先级顺序对sRNA进行遍历,没有比上任何注释信息的用unann表示(Burgeet al.,2013)。 由于 rRNAetc是由NCBI Genbank和Rfam 2个数据库比对所得,规定这2个数据库间的优先级为Genbank>Rfam11.0。
1.4 已知miRNA比对
将sRNA和miRBase数据库(http:∥www.mirbase.org/)中所有昆虫miRNAs进行比对,鉴定样本中保守miRNA,并预测保守miRNA的靶基因(Ambros, 2003; Ana&Sam, 2010,2013; Griffithsjoneset al.,2006,2008; Meyerset al.,2009)。
1.5 新miRNA预测
新miRNA是指在与miRBase数据库中所有昆虫miRNAs进行比对时,未注释上任何RNA且比对上基因组外显子反义链、内含子、基因间区的小RNA,通过选用软件mitp(miRNA identification and target prediction pipeline)筛选miRNA的生物特征得到的(Friedländeret al., 2008)。
1.6 miRNA差异表达分析
使用ExpDiff方法对miRNA进行差异表达分析,将野外采集的松墨天牛的miRNA库(NJ)和室内饲养的松墨天牛的miRNA库(LKY)的各组织进行比对。对所有数据进行归一化并对数转化。对归一化后的数据进行组间t检验,差异表达miRNA筛选条件:P-value≤0.05且fold change≥2(魏明辉等,2014)。
1.7 miRNA的靶基因预测及功能预测
使用miRanda软件对差异表达的miRNA进行生物信息学分析,预测靶基因(Hsuet al.,2008)。将靶基因投射到 Gene Ontology数据库(http:∥www.geneontology.org/),进行基因GO分析(Harriset al.,2004)。将靶基因与参考基因进行比较,选择靶基因中显著富集的几个GO功能条目,并筛选出与其显著相关的生物学功能。用KEGG(Kyoto Encyclopedia Genes Genomes)数据库(http:∥www.genome.ad.jp/kegg/)提供代谢通路信息进行Pathway富集分析,当Q-value≤0.05时,表示差异表达基因在该通路中显著富集(Kanehisaet al.,2000,2008)。
2 结果与分析
2.1 miRNA测序的结果统计
应用illumina测序平台完成野外松墨天牛的表皮(NJ-Ep)和中肠(NJ-Mg)与室内饲养的松墨天牛的表皮(LKY-Ep)和中肠(LKY-Mg)4个样品的miRNA测序。经分析,4个库的 clean reads中,LKY-Ep、NJ-Ep、LKY-Mg、NJ-Mg 的 GC 含量分别为40.53593%、41.26336%、41.62256%、41.69211%,GC含量位于35%~65%之间,说明测序质量好。测序长度集中分布在21~22 nt范围内,长度为22 nt的reads占总reads的比例最高。野外采集与室内饲养的松墨天牛的测序片段分布具有明显的相似性。
通过bowtie将sRNA定位到基因组上,4个库中得到的原始序列分别为 11842208、11528297、11852104、12118488个,过滤低质量(Q-value<30)和接头序列后,NJ-Ep样品中,高质量 reads为11692536个,clean reads为10976643个,占高质量reads的93.88%;NJ-Mg的样本中,高质量 reads为11383034个,clean reads为 10750830个,占高质量reads的94.45%;LKY-Ep的样本中,高质量的reads为11704237个,clean reads为11165380个,占高质量reads的95.40%;LKY-Mg的样本中,高质量的reads为 11965143个,clean reads为 11579370个,占高质量reads的96.78%。说明测序所获得的数据量大,测序读长质量高。将得到的所有有效数据比对光肩星天牛的基因组,发现分别有25.62%、25.99%、37.06%、33.45%的 unique sRNA 在 NJ-Ep、NJ-Mg、LKY-Ep、LKY-Mg中能定位到基因组上。
通过blast将sRNA与Genbank和Rfam数据库中的非编码 RNA进行比对,包括核糖体 RNA(rRNA)、核仁小RNA(snoRNA)、核内小RNA(snRNA)、转运RNA(tRNA)。筛选和去除其中的其他非编码RNA的干扰。其丰度分布如图1所示,可以看出实验室饲养的松墨天牛老熟幼虫的miRNA丰度大于野外松墨天牛。
2.2 保守miRNA的鉴定
将sRNA和miRBase数据库(http:∥www.mirbase.org/)中所有昆虫miRNAs进行比对,构建保守miRNA表达谱。
在 LKY-Ep、LKY-Mg、NJ-Ep、NJ-Mg 的样本中,鉴定到保守 miRNA 分别为 16、14、13、13个,表达量分别为 1509、2296、1301、1621。 从组织特异性来看,中肠比表皮的miRNA表达量高;从室内饲养与野外捕获的角度来看,室内饲养的松墨天牛miRNA表达量更高一些。
通过miRDeep2根据比对到基因组上sRNA的序列,识别保守miRNA的丰度和表达量情况。在表1中列出了在4个库中表达量最高的10个保守miRNA。
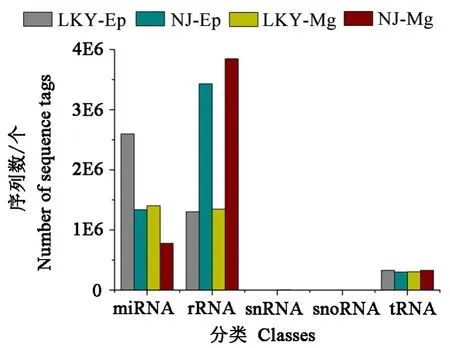
图1 室内饲养与野外采集的松墨天牛幼虫的小RNAs的分类图Fig.1 Distribution of different small RNA classes in indoor rearing and wild M.alternatus larvae
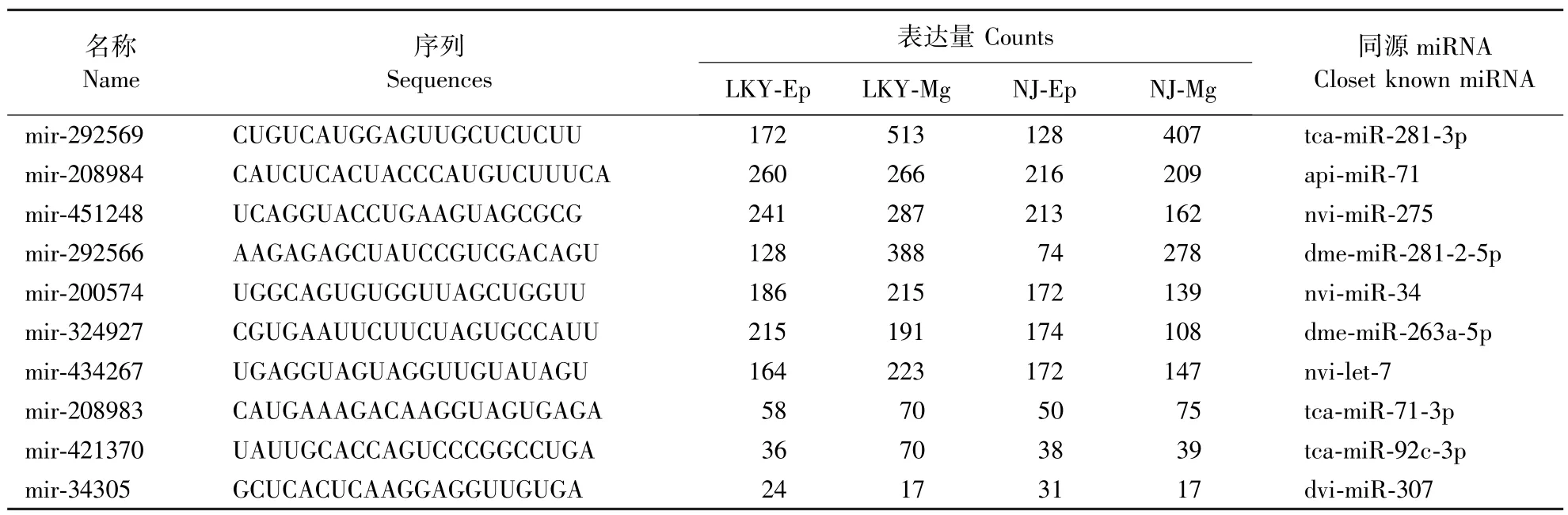
表1 松墨天牛幼虫的4个库中保守miRNA中表达量最高10个miRNA的序列和表达量Table 1 Sequences and abundance of top ten conserved miRNA candidates in four small RNA libraries of M.alternatus larvae
2.3 新miRNA的预测及表达量结果
在 LKY-Ep、LKY-Mg、NJ-Ep、NJ-Mg 的样本中,分别鉴定到 431、514、344、414 个新 miRNA,表达量分别为 3581、5050、2487、3217。 表 2中是从 4 个文库中筛选出表达量前10位的新miRNA。
2.4 保守miRNA的靶基因预测
用miRanda对保守miRNA进行靶基因预测,选取能量值低于-84 kj·mol-1,且得分值大于140的进行统计,可得出室内饲养的松墨天牛表皮(LKY-Ep)的16个保守miRNA对应16673个靶基因,中肠(LKY-Mg)的14个保守miRNA对应14451个靶基因。野外松墨天牛表皮(NJ-Ep)的13个保守miRNA对应13763个靶基因,中肠(NJ-Mg)的13个保守miRNA对应13763个靶基因。

表2 松墨天牛幼虫的4个小RNA文库中预测出的前10位新miRNA及其序列和表达量Table 2 Sequences and abundance of top ten predicted novel miRNA candidates in four small RNA libraries of M.alternatus larvae
2.5 差异表达的miRNA的靶基因功能预测
保守miRNA差异分析发现,分析到的保守miRNA无显著差异。故对所有的miRNA进行分析,在室内饲养后,中肠的miRNA表达量总体高于表皮,室内饲养的松墨天牛的表达量较高于野外松墨天牛。某些microRNA表达量与野外捕获的天牛有明显变化,如 novel-mir-62127、novel-mir-184731、novel-mir-290819明显上调,novel-mir-251851明显下调(图2)。
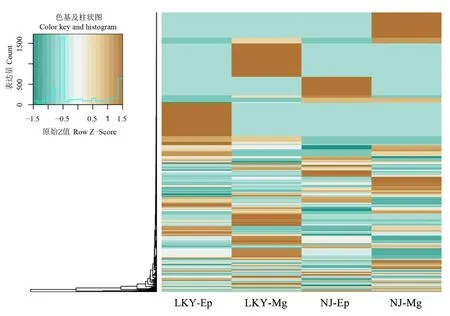
图2 松墨天牛幼虫4个库中差异表达miRNA聚类分析Fig.2 Cluster analysis of the expression profiles of verified miRNAs in 4 small RNA libraries of M.alternatus larvae
从图3中可以看出,这些差异表达miRNA靶向的基因功能大都相近,主要集中在氧化还原过程、细胞外组分、氧化还原酶活性3个分类单元。并且富集到很多代谢途径,如氨基糖、几丁质、含氨基葡萄糖的化合物、碳水化合物代谢途径等。说明室内饲养可能影响了天牛的代谢与形态结构,并且酶活性受到很大影响,如氧化还原酶、几丁质酶、水解酶、酰基辅酶A脱氢酶的活性等,可能是由于室内饲养的恒温条件与野外环境变化的温度导致。
KEGG通路富集分析发现(图4),室内饲养与野外天牛的表皮和中肠中,代谢水平发生了巨大的变化,如丙酸酯、甘油磷脂、脂肪酸、色氨酸代谢等,说明不同的生活环境对于天牛的代谢有重要影响。
3 讨论
本研究通过高通量测序分析鉴定室内饲养与野外捕获松墨天牛的miRNA,得到基因序列的注释信息。结果发现,从长度分布上可以看到,主要分布在21~23 nt,该长度符合miRNA长度分布模式(Lagos-Quintanaet al., 2001; Lauet al.,2001)。 并且在4个miRNA文库所有表达量结果中,室内饲养后的松墨天牛miRNA的表达量高于野外采集天牛,2组中肠的表达量高于表皮,此结果与宁静等(2018)的结果一致。这可能说明:从表观遗传的角度上,中肠相对表皮组织在冷适应方面响应更强烈,比表皮发挥更多的功能(Hunget al.,2000;Cristofolettiet al.,2003; Matthewset al.,2010)。 室内饲养的松墨天牛miRNA上调表达可能会通过抑制一些生理过程的基因,从而影响其代谢、发育过程。此外,由于本研究用光肩星天牛的基因组作为miRNA的鉴定依据,限制了比对的范围,导致鉴定到的总miRNA的数量较少。随着高通量测序以及生物信息学的迅猛发展,待松墨天牛基因组测序完成,对于松墨天牛miRNA组学的研究将会更加简便、全面。
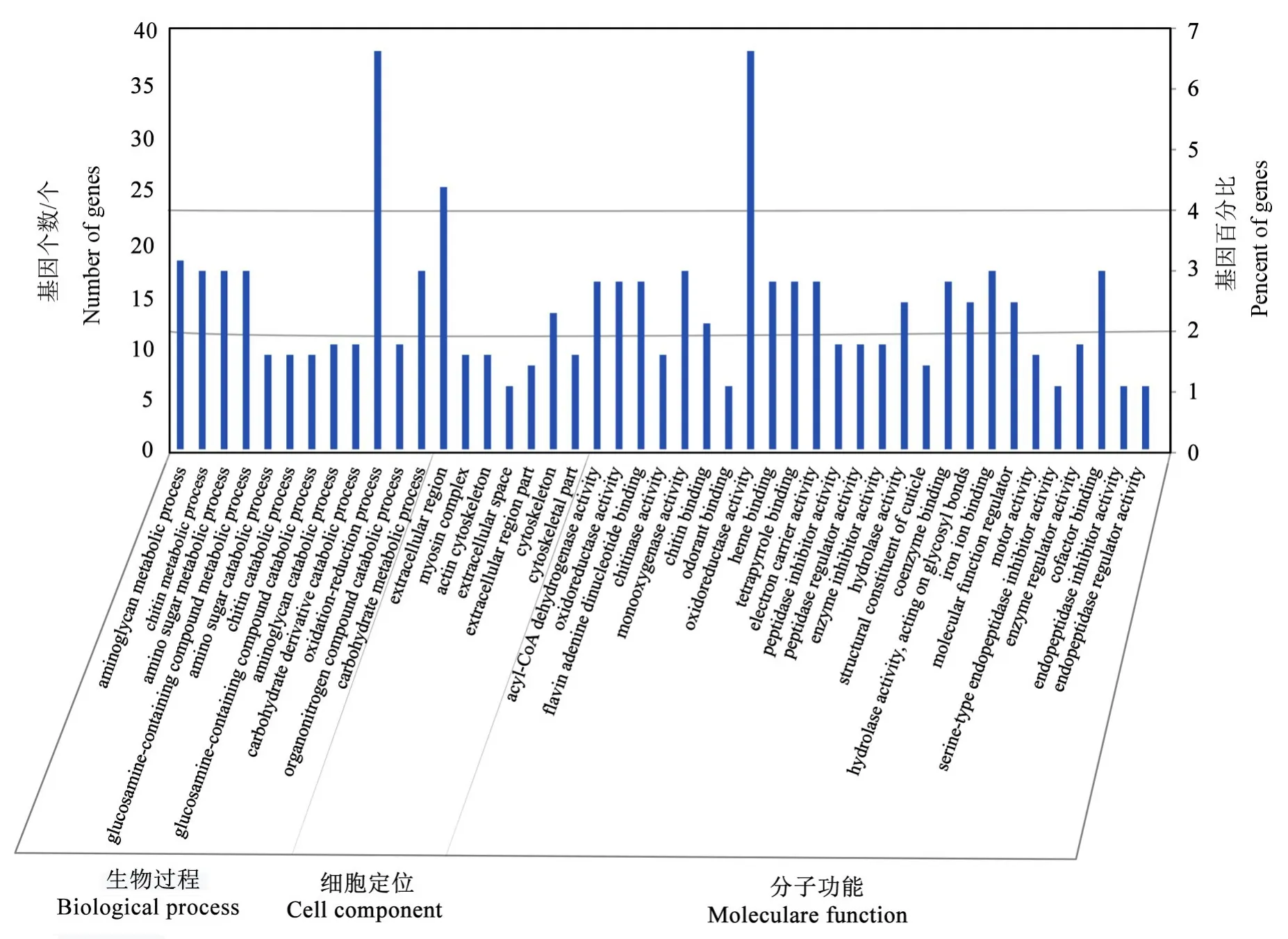
图3 松墨天牛幼虫差异表达miRNA靶基因的GO功能分类结果Fig.3 Result of GO function classification of target genes of differential expression miRNA in M.alternatus larvae
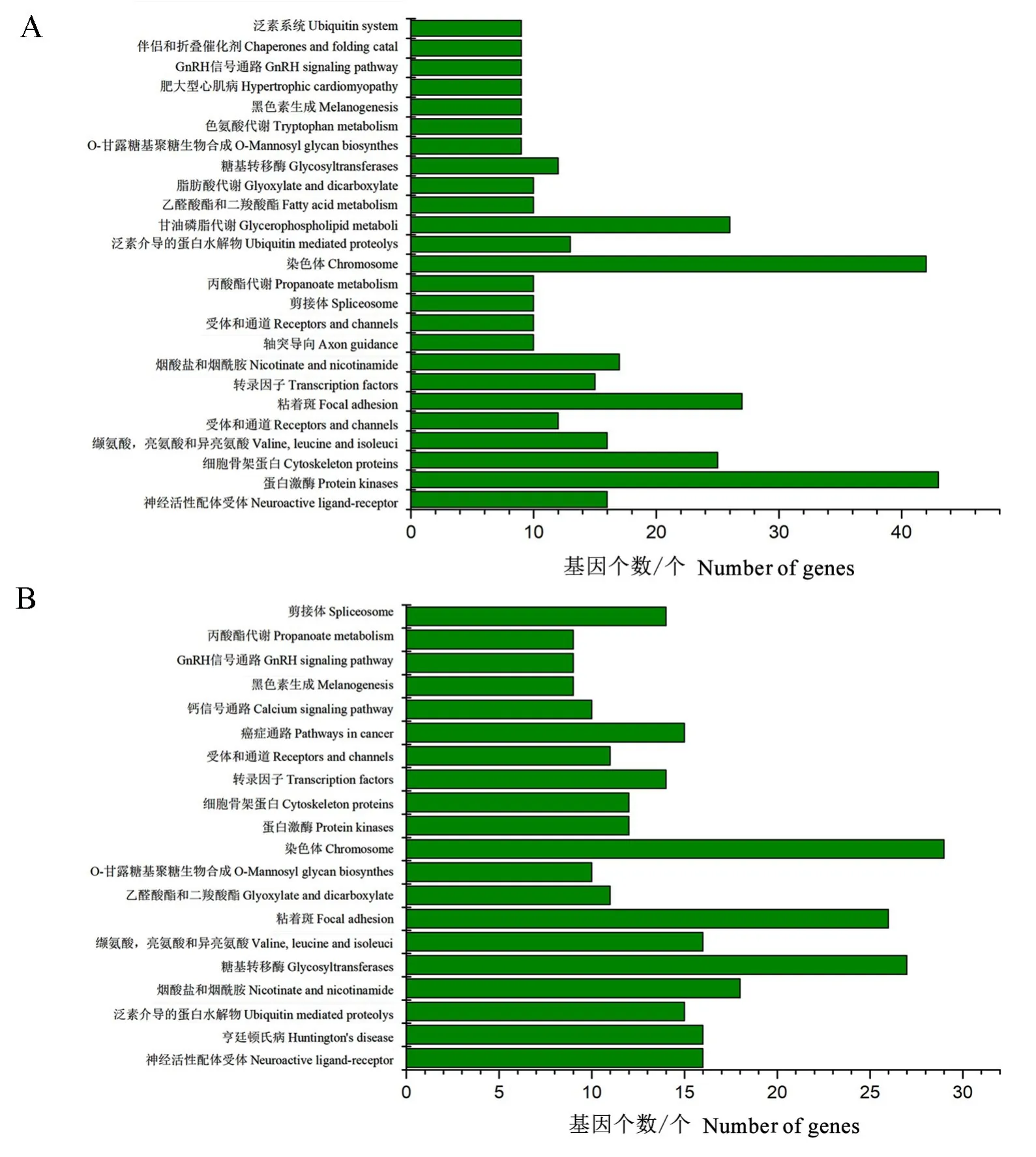
图4 差异表达miRNA靶基因的KEGG通路富集分析结果Fig.4 Results of the KEGG pathway enrichment analysis of target genes of differential expression miRNA
在室内饲养后,有些miRNA表达量有明显的变化,比如 novel-mir-62127、novel-mir-184731、novelmir-290819明显上调。由于分析得到的差异表达miRNA未在昆虫中注释,故比对到其他物种鉴定同源miRNA。novel-mir-62127比对到的同源miRNA为osa-miR166i-5p,与耐热性相关,可能由于实验室与野外环境的温度不同(Liuet al.,2017)。mir-62127等一系列的具有显著差异的miRNA,目前在昆虫中未有注释,这些松墨天牛的新miRNA可能调节新的靶标基因,具有新的功能。本研究只是一个初步的数据分析,有关基因的挖掘及其表达和功能验证还需进一步研究。
以往研究发现,室内饲养会很大程度影响松墨天牛的代谢以及形态结构的变化,会使其发育历期缩短,体型变小,产卵量减少(徐金华等,2009)。GO功能预测发现,差异基因很多与糖代谢途径相关,比如:氨基糖代谢途径、氨基糖分解代谢过程、几丁质代谢途径、几丁质分解代谢过程、含氨基葡萄糖的化合物代谢途径、碳水化合物代谢途径等代谢途径中有较多的富集,表明实验室饲养对松墨天牛的糖代谢有重要影响,这可能与信息素ascarosides的产生或者代谢相关。几丁质代谢途径、几丁质分解代谢过程的富集意味着在蜕皮时间上有所调控,如野外的松墨天牛幼虫是一年一代,从12月到来年的4月一直处于老熟幼虫期,5月才开始变态发育进行蜕皮,但是在室内恒温培养下发育蜕皮速度加快,一年为3~4代,因此,这2个信号途径有助于进一步探究其在蜕皮速度调控中的功能。在KEGG富集分析中也发现,室内饲养对脂肪酸代谢、甘油磷脂代谢等脂代谢相关过程有影响,这为后续实验提供了思路。利用室内miRNA抑制的代谢靶标基因进一步研究及开发代谢抑制剂或干扰剂,可扰乱松墨天牛代谢过程以达到防治目标。所以本研究对野外干扰剂的开发具有一定的指导意义。并且在KEGG通路中发现染色体的富集水平较高,室内外的温度差异可能是对染色体产生影响的主要原因(Bora&Soper,1971)。总体来说,室内饲养与野外捕获的松墨天牛存在一定的差异,希望本研究结果为后续挖掘两者不同表型的表观遗传调控机制及防治害虫研究提供一定的帮助。
致谢:江苏省林业科学院帮助采样,特此表示感谢!
猜你喜欢
农业灾害研究(2022年11期)2023-01-09
中国生物防治学报(2022年3期)2022-07-09
保健文汇(2022年1期)2022-06-06
绿色科技(2021年18期)2021-10-16
科技创新与应用(2021年7期)2021-02-04
中国森林病虫(2020年6期)2020-11-28
放射学实践(2020年2期)2020-03-13
绿色中国(2019年13期)2019-11-26
现代园艺(2017年21期)2018-01-03
现代园艺(2017年21期)2018-01-03
