
原发皮肤CD4+小/中等大T细胞淋巴组织增生性疾病临床病理特征及预后分析
2020-06-29 09:15黄雪洁王冠男赵武干崔美英张崇立李文才
临床与实验病理学杂志 2020年5期
黄雪洁,王冠男,赵武干,崔美英,张崇立,李文才
WHO(2016)淋巴造血系统肿瘤分类将“原发性皮肤CD4+小/中等大T细胞淋巴瘤”更名为“原发皮肤CD4+小/中等大T细胞淋巴组织增生性疾病(primary cutaneous CD4+small/medium T-cell lymphoproliferative disorder, CD4+PCSM-TCLD)”,但其仍做为暂定的分型[1]。该命名因其病灶多为局限性且预后较好,使其不再归为淋巴瘤,但其惰性的临床过程与组织病理学和分子特征之间不一致,使其在良恶性分类中存在争论。本文回顾性分析6例 CD4+PCSM-TCLD的临床特点和病理学特征并分析其预后,旨在提高临床及病理医师对该病的认识水平。
1 材料与方法
1.1 材料收集2015年6月~2019年10月郑州大学第一附属医院病理科诊断的6例CD4+PCSM-TCLD。所有病例均按WHO(2016)淋巴造血系统肿瘤分类诊断标准由两位副高以上淋巴瘤亚专科医师重新阅片、分类诊断。
1.2 方法
1.2.1免疫组化 采用免疫组化EnVision法染色。抗体:CD2、CD3、CD4、CD5、CD7、CD8、CD30、CXCL-13、CD21,均购自福州迈新公司;PD-1、CD10、BCL-6、CD68、Ki-67,均购自北京中杉金桥公司。结果判断:CD2、CD3、CD4、CD5、CD7、CD8、CD20、CD30阳性定位于细胞膜,PD-1、CXCL-13阳性定位于细胞质,BCL-6、Ki-67阳性定位于细胞核。
1.2.2EBER原位杂交 EBER试剂盒购自北京中杉金桥公司,具体操作步骤严格按试剂盒说明书进行,应用辣根过氧化物酶,DAB显色。以EBV阳性NK/T细胞淋巴瘤做为阳性对照,以PBS缓冲液替代杂交液或一抗作为阴性对照。结果判断:EBER阳性信号定位于细胞核,呈棕黄色。
1.2.3T细胞克隆性分析 DNA提取试剂盒为QIAamp DNA Mini Kit,PCR引物由美国Invivoscribe公司合成。(1)PCR扩增:按试剂盒说明书进行组织DNA的提取,分光光度计测其A值,PCR扩增检测提取的DNA质量,合格后行聚合酶链式反应扩增。TCR基因(TCRβ+TCRγ+TCRδ)检测采用BIOMED-2引物系统。(2)基因扫描:PCR反应体系为25 μL,PCR基因产物混合液经95 ℃变性10 min,冰水浴10 min,最后进行克隆性分析(ABI公司,ABI3500DX遗传分析仪)。(3)结果判读:单克隆性重排的判定为目标位置上位于高斯分布区域内出现明显突出、单一大小的条带,多克隆性重排的判定为收集到连续的多峰荧光信号。
2 结果
2.1 临床特点本组患者女性2例,男性4例,发病年龄19~53岁,中位年龄36.5岁,平均36.2岁。发病部位2例位于颈部,3例位于面部,1例位于枕部,均为单发结节/斑块,病程1~12个月,例3病变最大径达10 cm。有随访资料的5例患者均无肿瘤及自身免疫性疾病相关病史,无系统性症状,血常规、乳酸脱氢酶及β2微球蛋白无明显异常,淋巴结无受累。例2为门诊会诊患者,诊断后失访(表1)。
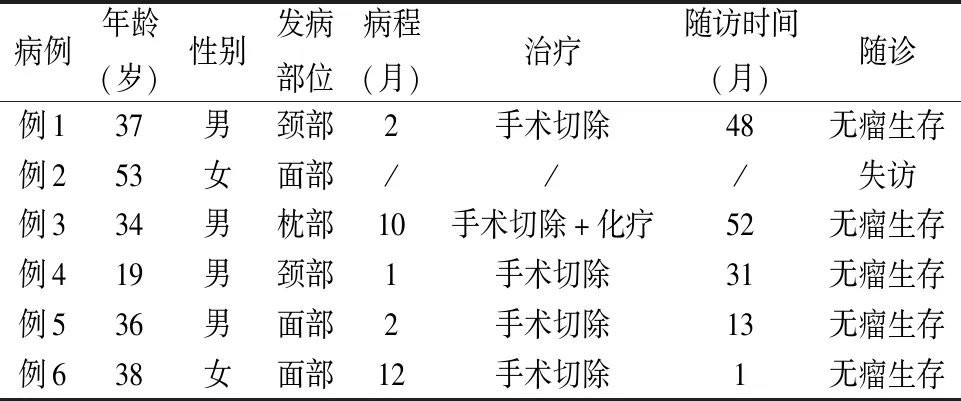
表1 CD4+PCSM-TCLD临床特点及随访
2.2 组织学特征低倍镜下见病变主要分布于真皮层,皮肤附属器可见破坏、减少,甚至消失,6例均累及部分皮下脂肪组织,表皮均未见受累。例1~5病变生长模式呈结节状和弥漫状,且病变浅层呈弥漫状分布,病变深层由于纤维组织分割呈模糊结节状分布(图1),例6仅表现为大小不一的结节状分布。肿瘤间质可见明显增生的小血管,部分有分支。高倍镜下见6例肿瘤细胞形态均小~中等大小,胞质少,细胞轻度异型,核型稍不规则,染色质细腻或略粗糙,核仁不明显,例1~5核分裂象罕见,例6偶见核分裂象。本组有5例(例1~5)病变背景中混杂较多B细胞、组织细胞及浆细胞等(图2),例6背景细胞较少。6例均可见散在分布的免疫母细胞样大细胞(5%~15%)。另外,例1可见明显上皮脚下延,例5可见由上皮样组织细胞组成的小肉芽肿结构(图3)。
2.3 免疫表型及原位杂交本组6例病变细胞均表达CD2,3例T细胞标记缺失,其中CD3、CD7各缺失1例(16.7%),CD5缺失3例(50%)。6例均CD4+(图4)/CD8-。滤泡辅助T细胞标记:5例(5/5)CD10均阴性,5例(5/5)PD-1局部可见围绕大细胞周围的环状阳性(图5),5例(5/5)BCL-6阳性(2例阳性,3例部分阳性),2例(2/5)CXCL13阳性。6例均有CD20阳性细胞散在或结节状分布,例6中CD20阳性细胞主要分布于病灶结节的外围。CD30为大细胞散在阳性(5%~15%)。6例CD21均显示无FDC网。例1~5的Ki-67增殖指数为5%~40%,例6的Ki-67增殖指数为80%(表2,图6)。EBER原位杂交均为阴性(6/6)。

图1 病变位于真皮层,侵及部分皮下脂肪组织,弥漫状及结节状分布,可见上皮脚下延 图2 病变主要由中小淋巴细胞组成,其间混杂多形性炎细胞,间质血管增生 图3 例5可见由上皮样组织细胞组成的小肉芽肿结构 图4 CD4呈阳性,EnVision法 图5 PD-1大细胞周围花环状阳性,EnVision法 图6 例6的Ki-67增殖指数80%,EnVision法

表2 CD4+ PCSM-TCLD的免疫表型
*例4为会诊病例,无存档蜡块未行滤泡辅助T细胞标记检测,查阅病史、影像资料、实验室检查、随访并重新阅片,结合EBER和TCR基因重排均支持CD4+PCSM-TCLD诊断
2.4 分子检测6例经TCR基因重排检测均提示存在单克隆性增生的T细胞群(图7)。
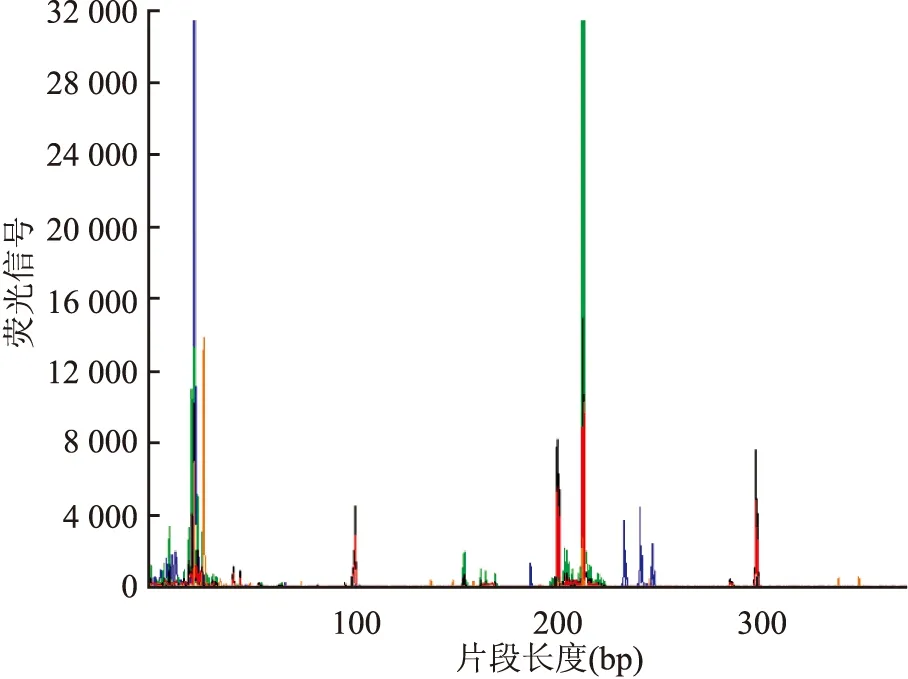
图7 TCR基因呈单克隆性重排
2.5 治疗及随访本组6例患者中4例仅接受了手术局部切除治疗,1例术后接受2个周期CHOP(表柔比星+环磷酰胺+长春新碱+泼尼松)+2个周期的FC(环磷酰胺+氟达拉宾)方案化疗,1例失访。随访时间1~52个月,中位随访时间31个月,有随访资料的5例患者均无瘤生存。
3 讨论
CD4+PCSM-TCLD属于原发皮肤的少见淋巴组织增生性疾病,占原发性皮肤淋巴瘤的2%~3%,发病年龄3~90岁,但多见于中老年人,中位年龄50~60岁,无明显性别优势。CD4+PCSM-TCLD临床病程较短,常以头颈部皮肤的单发无痛性结节/斑块为症状,缺乏系统性表现,实验室检查无异常,多灶性病变亦有报道[2],病变直径最大者可达14 cm[3]。本组6例患者发病年龄小,男性较多,病灶最大径为10 cm,余临床资料与既往报道相符。
CD4+PCSM-TCLD病变位于真皮层,累及皮下脂肪层,但真皮浅层无浸润带,表皮无侵犯,细胞成分多样,主要由中小淋巴细胞组成,呈结节状或弥漫性生长,其间可见多种炎细胞浸润[2-4]。本组组织学形态与文献报道基本一致,但所有病例均可见免疫母细胞样大细胞数量不一、散在分布,且所占比例较少(<15%),有1例可见明显肉芽肿形成,1例可见明显上皮脚下延,上述组织学表现以往鲜有学者关注,但未见详细表述。因此,肉芽肿形成和上皮脚下延拓展了CD4+PCSM-TCLD的组织病理形态学谱系,使其形态学表现更为复杂多变,而免疫母细胞样大细胞的比例高低是否与预后有关,有待于扩大样本量进行评估。CD4+PCSM-TCLD表达广谱T细胞标记,会出现个别抗原丢失,以CD5和CD7多见[5],CD2丢失也有报道[3]。 CD4+PCSM-TCLD为滤泡辅助T细胞来源,因此抗原PD-1、BCL-6、CXCL-13有不同程度表达,但CD10均阴性,Krenács等[6]检测CXCR5也均未表达。值得注意的是,Rodriguez-Pinilla等[7]运用免疫组化双染显示病变细胞在CD30/OCT-2+B细胞周围形成玫瑰花结,对突出病变细胞的排列方式非常有帮助。CD4+PCSM-TCLD的Ki-67增殖指数多为5%~40%,EBER原位杂交阴性,TCR重排单克隆阳性率高于60%。本组患者有CD3、CD5及CD7表达的丢失,其中例3出现CD5和CD7的共缺失,例6出现CD3和CD5共缺失。例6除T抗原丢失外, Ki-67增殖指数高达80%,其与外周T细胞淋巴瘤的鉴别存在一定困难和争议,但该患者临床呈惰性病程,无系统性症状,且影像学检查未见明显淋巴结肿大和结外表现,组织形态学异型性不明显,因此诊断暂将其放入CD4+PCSM-TCLD,但是否会转化为外周T细胞淋巴瘤有待进一步随访分析。
CD4+PCSM-TCLD需与发生于皮肤的其它淋巴组织增生性疾病/淋巴瘤鉴别。(1)淋巴组织假瘤样增生:细胞缺乏异型性,免疫表型基本无T抗原的丢失,NFATc1表现为细胞质着色[8],PD-1多为散在表达模式[9],分子检测为多克隆性。CD4+PCSM-TCLD在文献报道及本组病例中均显示混杂较多B细胞,甚至肉芽肿的形成,为鉴别诊断增加了困难,TCR基因重排有助于两者鉴别[10]。(2)蕈样霉菌病的结节期:临床病程及皮损情况有助于鉴别。(3)原发性皮肤滤泡辅助T细胞淋巴瘤:临床上多为系统性病变,侵袭性病程,且多数病例具有CD10的表达。(4)外周T细胞淋巴瘤,非特指型:属高侵袭性淋巴瘤,细胞异型性更大,Ki-67增殖指数较高,广谱T细胞标记丢失更加常见,多不表达PD-1。
CD4+PCSM-TCLD临床多呈惰性过程,5年生存率超过90%,治疗上主要采取单纯切除,个别报道活检后病灶甚至可自发消退[11]。国内文献报道CD4+PCSM-TCLD均为单发病灶,且表现较好的预后[12-13],但国外文献Garcia-Herrera等[14]报道5例患者死亡,预后不良指标包括:病变直径大于5 cm、Ki-67增殖指数较高、背景CD8+细胞减少等。本组患者中例3出现最大径达10 cm的病灶,例6的Ki-67增殖指数达80%,但例3及例6分别随访52个月和1个月,目前患者均无瘤生存。虽然本组2例可见上述预后不良因素,但随访结果较好,与文献报道并不一致。例3患者预后较好,可能与化疗相关。因此,CD4+PCSM-TCLD中预后不良因素有待扩大样本量,并延长随访时间进一步分析。
总之,CD4+PCSM-TCLD作为原发皮肤的少见淋巴组织增生性疾病,具有独特的临床表现和病理学特征。本组患者有免疫母细胞样大细胞、上皮脚下延和肉芽肿形成,拓展了CD4+PCSM-TCLD的组织形态学谱系,使其形态学表现更为复杂多变。此外,本组病灶较大、Ki-67增殖指数较高的患者在随访过程中依然取得了较好的预后,因此该病的预后不良因素有待进一步讨论。临床及病理医师也应该加深对该病的认识水平,以便给予适当的治疗方案。
猜你喜欢
传染病信息(2022年3期)2022-07-15
世界最新医学信息文摘(2021年12期)2021-06-09
石油化工自动化(2020年1期)2020-03-05
通信技术(2019年8期)2019-09-03
中国循证儿科杂志(2019年2期)2019-06-04
家庭医药·快乐养生(2019年2期)2019-03-04
中华老年多器官疾病杂志(2016年9期)2016-04-28
听力学及言语疾病杂志(2015年5期)2015-12-24
磁共振成像(2015年5期)2015-12-23
天津医科大学学报(2015年3期)2015-06-05
