
Ce-Zr-Al-O催化剂催化烟梗热解的分析
2020-06-29 04:12李文斌郑云武李水荣刘志华刘灿郑志锋
化工进展 2020年6期
李文斌,郑云武,李水荣,刘志华,刘灿,郑志锋,
(1 云南省烟草化学重点实验室,云南昆明650000;2 林业生物质资源高效利用技术国家地方联合工程研究中心,西南地区生物基功能材料创新示范平台,西南地区林业生物质资源高效利用国家林业和草原局重点实验室,西南林业大学材料科学与工程学院,云南昆明650224;3 厦门市现代农业生物质高值化技术重点实验室(厦门大学),福建省生物质高值化技术工程研究中心(厦门大学),厦门大学能源学院,福建厦门361102)
化石燃料储量随着时间的推移变得越来越有限,而且获取成本增加,同时化石燃料的消耗也影响了气候变化[1]。生物质热解是一种有效的手段,通过从农林业废弃物中获取燃料和定向获得高价值的化学品,从而达到成本的最优化,同时将环境污染降至最低[2]。中国是世界上最大的烟草生产国和消费国,年烟草产量为400~500 万吨,每年产生约200 万吨烟草废物,包括烟叶碎片和烟梗[3]。烟梗作为烟草行业的副产物仅有极少部分作为原料加入工业卷烟,且不能采用一般的堆肥和填埋手段处理,因此,通过生物质热解技术制备生物油,从中提取高附加值化学品是一个有前景的研究方向。目前,烟草行业的大部分研究基于烟叶开展,Akalin等[4-5]利用固定床研究温度和催化剂对烟草残渣热解的影响,发现热解产生的生物油含有许多含氮的化合物,其次还包括酸、酯、烷烃和酚类化合物。Mitsui 等[6]利用热解气相色谱-质谱联用仪(Py-GC//MS)系统分析了烟草快速热解过程中产生的生物油的化学成分,发现烟叶和香烟烟雾的气味是由氨基酸和糖类的美拉德反应(Maillard reaction)产生。Yan 等[7]使用流化床反应器对烟叶(TL)和烟梗(TS)进行了热解研究,目标是制备含有芳香化合物的生物油或用作液体燃料。然而,烟梗热解转化研究报道较少,尤其是催化热解定向获得具有较高附加值的生物油和化学品。CeO2具有优异的氧化还原性能,高氧空位浓度,多极化多孔结构,可为催化剂上的氧扩散提供额外的通道[8],但是对水蒸气敏感且在高温条件下易发生烧结[9]。因此,提高烟梗在高温下的热稳定性显得尤为重要。有文献指出,在CeO2中掺杂Zr4+,形成CexZr1-x固溶体(以下简称CZ),可以改善催化剂的体相特征,在增加热稳定性的同时也显著提高了它的储氧能力[10-12]。Al2O3作为一种高熔点且热稳定性极好的材料,酸性和碱性中心分布在其表面,在催化领域已经被广泛利用。Ce-Zr-Al-O 催化剂(以下简称CZA)是在CZ 中加入杂原子Al,这样既可以获得Al2O3的高比表面积,也具有优良的储存和释放氧的能力[13]。
目前,烟梗的研究主要用于提取烟碱、制造纤维复合材料和生物炭等,对于慢速热解催化热解过程和催化产物的分析研究较少。基于此,本文利用废弃烟梗,采用高温慢速催化热解技术研究烟梗催化热解转化产物的分布和热解过程,弥补目前在烟梗慢速催化热解制备高附加值化学品的不足,以期为烟草废弃物及生物质的高效转化和定向催化转化制备高附加值化学品提供一定的理论依据。
1 实验部分
1.1 材料及试剂
烟梗,云南中烟集团提供。自然风干后粉碎过筛,选择粒径尺寸在0.075~0.180mm之间的粉末,置于(105±2)℃烘箱中干燥24h,然后收集、储存、备用。烟梗的C、H、O、N元素分析采用德国元素公司生产的EA 1108型号元素分析仪测定。烟梗的工业分析参考专利[14]介绍的方法测定。烟梗的热值采用式(1)进行计算[15]。

式中,C、H、O、N、S、Ash分别表示烟梗中各元素及灰分的质量分数。测试结果如表1 所示,从表中可以看出烟梗元素组成中硫、氮含量相对较低,C/H 比值高说明其可以作为清洁能源的来源,挥发分含量高使得烟梗具备良好的催化转化的特性。六水合硝酸铈[Ce(NO3)3·6H2O,99.95%金属基]、九水合硝酸铝[Al(NO3)3·9H2O,99.99%金属基],上海阿拉丁生化科技股份有限公司。氯氧化锆(ZrOCl2·8H2O,99.9%金属基),上海麦克林生化科技有限公司。氨水(AR,25%~28%),重庆川江化学试剂厂。氮气(99.99%),厦门市林德气体有限公司。
1.2 烟梗热重分析实验
利用NETZSCH TG 209 F3型热重分析仪研究烟梗挥发分析出区间变化及热解特性。称取约10.0 mg 样品于铝坩埚中,以20K/min 的升温速率升至800℃,高纯氦气(30mL/min)为保护气。即可得热解失重曲线(TG)、微商热重曲线(DTG)。
1.3 催化剂的制备
CeO2的制备:将一定量的Ce(NO3)3·6H2O 固体溶于100mL 蒸馏水,向其逐滴加入氨水至混合液pH≥10,磁力搅拌3h后静置24h。移除上清液后抽滤剩余固液混合物并洗涤至滤液呈中性,固体物质于80℃烘箱中干燥12h 后取出置于550℃马弗炉中焙烧3h,即得CeO2催化剂。CZA(Ce∶Zr∶Al 的摩尔比为0.8∶0.1∶0.1)的制备:将一定量的Ce(NO3)3·6H2O、ZrOCl2·8H2O和Al(NO3)3·9H2O 溶于烧杯中,向其逐滴加入氨水至溶液pH≥10,浸渍搅拌3h后静置24h。移除上清液后抽滤至溶液呈中性,固体物质于80℃烘箱中干燥12h 后取出置于550℃马弗炉中焙烧3h。即得CZA 固溶体催化剂。CZ(Ce∶Zr 的摩尔比为0.8∶0.2)制备同上。
1.4 催化剂的表征
利用物理吸附仪(ASAP 2020 PLUS HD88,Micromeritics,USA)研究催化剂的比表面积及孔径参数。将约0.1g 样品于350℃真空下脱气3h,N2为吸附剂,用于计算催化剂表面积和孔体积,包括催化剂的比表面积、孔径参数、氮气吸附-脱附等温曲线及孔径分布图。使用冷场发射型透射电子显微镜(FE-SEM,JSMM-6701F,JEOL,Led)和扫描电子显微镜(TM3000 Tabletop Microscope)观察样品的形貌特征。使用X衍射仪(Ultimaiv,Rigaku)对样品进行X 射线衍射(XRD),扫描范围为5°~85°,扫描台阶宽度为0.02°,扫描速度为8°/min,波长为1.5406nm,扫描电压和电流分别为40kV 和40mA,狭缝为0.3mm。催化剂表面酸性分布通过NH3程序升温脱附实验(NH3-TPD)在Micromeritics AutochemⅡ2920 固定床上进行,在石英管中放置约0.1g 催化剂,300℃下恒温加热30min 除去表面吸附的水和易挥发性杂质后降温至100℃,然后用5.0%氨(NH3/He)吸附30min,切换气路为氦气,以10℃/min速率从100℃升至800℃。X射线光电子能谱(XPS)测量在室温下进行,使用Kα-X 射线光电子能谱仪(Thermo Fisher Scientific Co.,USA)测试,X射线束为100W,直径为200mm,激光扫描面积为2mm×0.4mm。热重分析采用德国耐驰公司的NETZSCH TG 209 F3 型热重分析仪,以10K/min的升温速率从50℃升至800℃,氮气气氛保护,气流量为30mL/min,进样量约10.0mg。
1.5 烟梗催化热解实验及产物分析
固定床热解试验装置由程序升温控制,升温模式为慢速升温。石英材质热解反应管分为热解段和催化段(由控温装置分别控制加热),且自上而下依次放入烟梗粉末、绝干玻璃纤维、催化剂和玻璃纤维(其中,烟梗与催化剂质量比为1∶2)。试验前对热解反应管路进行气体冲洗,氮气吹扫约10min,待整个热解管路为氮气氛围时开始加热。热解段以20℃/min 的升温速率升到500℃并保持30min,催化段温度在热解之前升温至500℃。试验过程中,冷凝器中的热解产物在异丙醇与甘油的混合液(-20℃)条件下收集并用甲醇溶液稀释。

表1 烟梗的元素分析和工业分析(质量分数)
收集到的热解产物利用气相色谱-质谱联用仪(GC-MS,SHIMADZU,GCMS-QP 2010PLUS) 检测其主要组分。以高纯氦气作为载气,载气流量为3.0mL/min;采用分流模式且分流比为60∶1;进样口温度为230℃。色谱柱为Rtx-5MS(30m×0.25mm×0.25μm),柱箱内程序升温为:50℃保持2min后以5℃/min升高至260℃。质谱仪采用电子轰击离子源(EI),离子源温度为230℃,电子能量为70eV,质谱采集范围m/z=45~500。参考标准软件NIST14和NIST14s 谱库以及本文作者课题组的相关研究结果[15-17]对样品进行检索分析,采用面积归一化法(相对峰面积,%)计算产品产率。
2 结果与分析
2.1 烟梗的TG-DTG分析
烟梗的TG 和DTG 如图1 所示,其热解过程大概分为6 个失重段:①50~120℃,②120~180℃,③180~230℃,④230~280℃,⑤280~360℃,⑥360~560℃。第①阶段的失重峰主要是由于烟梗表面的结晶水蒸发所引起,质量损失大约为4%;第②和③阶段是热解初始阶段,质量损失大约为15%,损失的质量主要是一些小分子化合物的释放,包括葡萄糖、果糖、尼古丁和一部分大分子挥发分物质的分解,以及半纤维素和部分纤维素的分解。第④~⑥阶段的质量损失峰最大达42%且温度范围最广,主要是纤维素、半纤维素、糖类、木质素和果胶的分解[18]。基于热重分析结果以及本文作者课题组之前的相关研究结果[15-17],选定烟梗的热解温度和催化温度均为500℃。

图1 烟梗的TG和DTG曲线
2.2 催化剂表征结果
2.2.1 N2吸附-脱附表征
表2所示为催化剂比表面积、孔容以及平均孔径的变化状况。由表可知,CeO2比表面积(54.1m2/g)和孔容(0.0982cm³/g)较小,且具有一定的微孔结构。Zr 原子的加入,催化剂的比表面积与平均孔径变化不大,而Al 原子的加入使得催化剂的比表面积与累积孔容显著增大,但平均孔径有所降低,这与仝姗等[19]的研究结果一致。N2吸附-脱附等温曲线及孔径分布如图2 所示,由图可得,三种催化剂均为H4滞后环,根据IUPAC 定义为Ⅳ类型曲线,均属于介孔催化剂且介孔孔道较为规则。在相对分压0.6~0.9范围内,没有出现明显的N2毛细凝聚,表明随着Zr、Al 原子的加入,催化剂的规则程度并没有发生变化。由图2(b)可以看出,3 种催化剂均具有双峰分布,孔径主要在3~4nm 和4~10nm,同时也可得出Zr、Al 原子的加入,双峰分布程度均有所增加。

表2 催化剂的孔结构性质
2.2.2 X射线衍射(XRD)表征

图2 催化剂的N2吸附-脱附等温曲线及孔径分布曲线
图3 所示为CeO2、CZ 和CZA 催化剂的XRD 谱图。由图可知,CeO2出现了28.58°的特征衍射峰,表明CeO2的晶相结构为立方晶相(标准立方晶相的特征衍射峰为28.4°)[20]。CZ、CZA和CeO2相比,特征衍射峰的位置并没有发生变化,说明Zr 原子和Al 原子的加入并没有破坏CeO2的晶型结构,但是CZA 的衍射强度有所降低以及峰形宽化,说明Al 原子的加入抑制了Ce 原子与Zr 原子的接触,起到一定的扩散阻碍作用,致其晶体粒径减小[21]。另外,根据谢尔乐公式计算得出CeO2、CZ、CZA 的平均晶粒尺寸分别为14.3nm、12.9nm和6.3nm,这与TEM 所得的结果一致。CZ 谱图中ZrO2的衍射峰强度很低以及CZA 谱图中没有检测到Al2O3的特征衍射峰,说明ZrO2和Al2O3高度分散在CeO2表面,另一方面也是由于二者的含量较低所致。
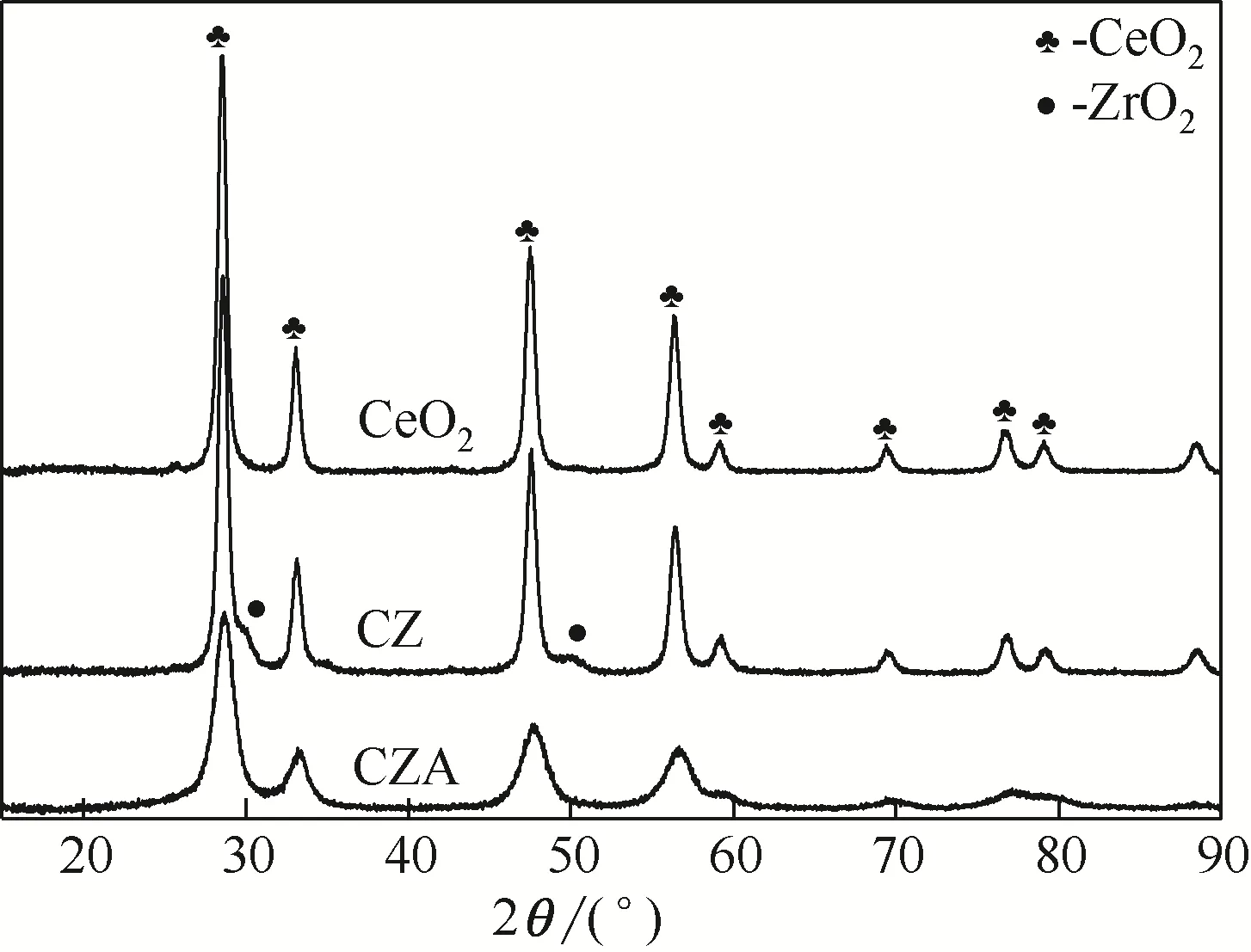
图3 CeO2、CZ和CZA催化剂样品的XRD图
2.2.3 程序升温脱附(NH3-TPD)表征
NH3-TPD 测定催化剂表面酸性,并对其进行拟合处理以不同温度下脱附峰的相对面积(SA为弱酸中心面积、SB为中强酸中心面积、SC为强酸中心面积)代表该条件下的酸量,结果如图4 所示。100~200℃的NH3吸附下的脱附峰为弱酸活性中心,200~350℃的脱附峰为中强酸活性中心,400~550℃的脱附峰为强酸活性中心[22]。CeO2表面的酸性主要是弱酸和强酸,且强酸的酸量较大。随着Zr 原子的加入,CZ 的弱酸显著增强且温度向高温方向偏移,同时中强酸活性位点增加,而强酸位点显著降低且向高温方向移动,说明Zr 原子可以消除CeO2的部分强酸性中心,增加弱酸中心和中强酸活性中心。于CZ 相比,CZA 表面的弱酸持续增加,中强酸和强酸含量没有明显变化,但中强酸酸性位向低温方向移动。
2.2.4 扫描电子显微镜(SEM)和场发射电子显微镜(TEM)表征
于是,我跟怡香院的老鸨建议,咱们举办一个名媛诗赛吧。邀请社交界的名媛和秦淮八艳PK诗歌,请大才子做评委,欢迎围观,不仅能打响姐妹们的名号,诗歌版权归咱们,还可以出诗文集,可以一版二版三版……银子那是哗啦啦的多。作为一个有经济头脑的老鸨,她很快欢欣鼓舞,并连续三天用看摇钱树的眼光关注我。
图5所示为3种催化剂的的TEM和SEM图。由TEM 图可知,CeO2晶体粒径的平均纳米尺寸为10~25nm,Zr原子的引入使得CZ晶体粒径的平均纳米尺寸降到13~19nm,说明掺杂Zr 原子抑制了CeO2晶体的增长。与CZ 相比,CZA 催化剂的晶体粒径更低,大部分晶体粒径在5nm 左右,说明Al原子在CZA 形成过程中将Ce 原子与Zr 原子的结合,起到一定的阻碍作用,致其晶体粒径减小。SEM 图可以看出,CeO2具有明显的立方体结构,晶体粒径不均一,结构具有多面性。CZ 和CZA 催化剂的颗粒形态与CeO2具有相似性,大多数颗粒趋向于立方形(尺寸为5~35μm),没有有固定的形状。

图4 CeO2、CZ和CZA催化剂样品的NH3-TPD图
2.2.5 X光射线电子能谱(XPS)分析
图6(a)所示为催化剂的全扫描XPS 分析谱图。由图可得,Ce3d能谱峰强度依次为:CeO2>CZA>CZ,说明Zr原子的加入抑制了Ce原子的聚集。其中,Ce4+的结合能由916.0eV 到916.7eV 再到917.1eV,向高结合能方向迁移,说明Zr 原子和Al原子的加入影响了Ce的化学环境以及Ce与Al2O3之间具有一定的协同作用[23]。依据谱图计算得出的表面原子比,催化剂表面氧原子占比约为30%。其中,CZA表面的氧含量高于CZ,CZ和CZA催化剂的Ce/Zr 原子比皆小于理论值4 和8,但CZA 的Ce/Zr原子比大于CZ,说明Al原子的加入导致Zr原子在催化剂表面发生富集,形成了“壳-核”结构,这与Fan 等[24]的研究结果一致。图6(b)所示为三种催化剂Ce3d 的XPS 谱图。Ce4+对应的特征峰为900.1eV、906.8eV、916.0eV、881.9eV、888.5eV和897.7eV,Ce3+对应的特征峰为903.4eV 和884.8eV。催化剂上氧空位的多少取决于Ce3+的数量,Zr、Al原子的加入促进了CZ 和CZA 表面Ce4+向Ce3+转化,氧空位数量增加,催化剂的储放氧能力增强。

图5 催化剂样品的TEM和SEM图

图6 催化剂的XPS谱图
2.3 烟梗催化热解
烟梗热解和催化温度均为500℃,原料与催化剂的质量比为1∶2,研究不同催化剂条件下的催化转化产物分布。检测到的化合物分为以下10类:酸类、醇类、酮类、酯类、糖类、酚类、含氮类、呋喃类、碳氢化合物和其他类。
由图7(a)可知,CeO2催化后酸类含量增加了0.72%,烟梗直接热解以及CZ 和CZA 催化后均没有检测到酸类化合物。纤维素、半纤维素和木质素的热分解均可产生酸类,但主要是由半纤维素解聚及脱水生成,而烟梗半纤维素含量很低,产生的酸含量很低,仪器并没有检测到。醇类主要来源于烟梗木质素中的脂肪族醇羟基结构,该结构在催化热解过程中断裂形成醇类化合物。CeO2和CZA 降低了醇类的含量,CZ 对醇类影响不大,可能是CeO2具有相对较小的比表面积,在催化热解过程中阻碍了羟醛缩合反应的发生,降低了醇类化合物的含量[25]。酯类在烟梗热解产物中占比较大,达7.48%,主要为3-甲氧基-2,4,5-三苯甲酸十一酯。催化剂的加入均抑制了酯类化合物的形成,尤其是CZA催化下,酯类化合物消失。这可能是因为CZA 催化剂表面的活性氧传递速率太快,不易缩合形成酯类物质。同时也有研究表明,醛和酯可以通过二次聚合转化为酮类化合物[26-27]。
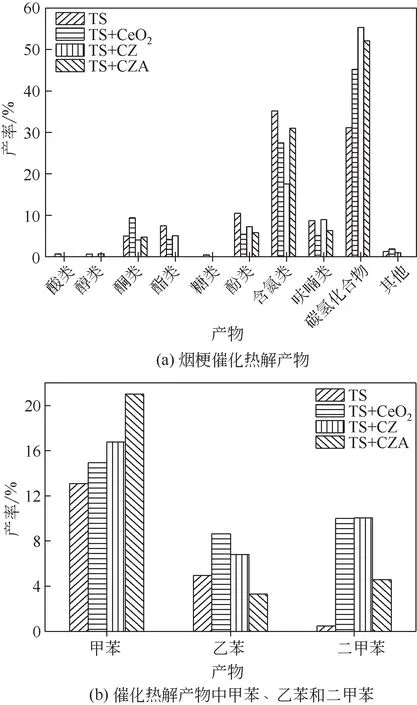
图7 烟梗催化热解产物分布以及催化热解产物中甲苯、乙苯和二甲苯分布
烟梗热解产物中酮类含量较低,主要为环戊酮和环己酮,一般为酸类或非酸类含氧化合物转化而来。CeO2提高了酮类含量,CZ 和CZA 对其影响不明显。CeO2可以在表面和孔道内通过形成金属羧酸盐中间体促进酮基化反应,而掺杂Zr、Al 原子后,这种特性有所降低[32]。此外,CeO2表面的强酸性活性位点高于CZ 和CZA,CeO2表面活性氧迁移较快,增强了氧化还原反应的性能,促进了热解产物的酮基化反应[33]。
酚类源于烟梗木质素裂解,主要为苯酚、烷基酚和烷氧基酚,烟梗热解产物中酚类可达10.47%,主要为4-乙基-苯酚、苯酚和2-甲基苯酚,催化剂的加入均降低了酚类总含量。但是,CeO2、CZ 和CZA提高了苯酚的含量而降低了烷氧基酚的含量且在CZA 作用下烷氧基酚完全消失,说明Zr、Al 原子的参与下,烷氧基酚被转化成其他的物质,这与Wang等[34]的研究结果相似。

碳氢化合物主要包含烷烃、烯烃和芳香烃,其中芳香烃占比最大。苯、甲苯、乙苯和二甲苯等轻质芳香烃可通过生物质定向催化热解制得,研究表明,HZSM-5 分子筛具有较好的耐酸性和耐热性,优异的选择性裂解和异构化性能,在生物质催化热解制备芳烃中被广泛使用[17]。但HZSM-5的微孔本质限制了大分子含氧挥发物的扩散,结焦率较高,催化剂表面易积炭失活。而本文的催化剂可以很好地解决催化剂表面的积炭问题。图7(b)所示为不同催化剂下甲苯、乙苯和二甲苯的变化趋势。甲苯在催化热解过程中一直呈现出增加的趋势,从13.08%分别增加到14.93%(CeO2)、16.75%(CZ)和21.01%(CZA),说明改性后的催化剂对甲苯的选择性更高。乙苯呈现出先增加后降低的趋势,在CeO2和CZ作用下分别达到8.64%和6.82%,但CZA作用下却抑制了乙苯的生成。二甲苯在烟梗热解产物中含量很低,催化剂加入均促进其生成,CeO2、CZ 和CZA 作用下,二甲苯含量分别达到9.99%、10.04%和4.58%。
2.4 催化剂的重复性和再生性
图8(a)所示为烟梗在CZA 催化剂连续使用3 次(用TS+CZA-x 表示,x=1,2,3)及焙烧再生后的催化热解产物分布。由图8(a)可知,催化剂使用次数的增导致产物中醇类、酮类、酚类和呋喃类含量有所增加,酸类、酯类和糖类未发生明显变化。催化剂使用次数的增加,酮类化合物相对含量持续增加,从4.75%到12.92%再增加到13.37%,呋喃类化合物也呈现出增加的趋势,从6.34%到8.36%再到8.65%,而碳氢化合物的相对含量择呈现出降低的趋势,从52%(一次使用)到49.34%(二次使用)再到43.66%(三次使用)。三次使用后的CZA经过简单焙烧后,活性基本恢复,且碳氢化合物含量达到52.1%。图8(b)所示为CZA 催化剂重复性测试前后的XRD 谱图(使用3次),晶型没有发生显著的变化且没有出现明显的积炭特征衍射峰(2θ=26.7°),表明催化剂表面并没有出现明显的积炭效应。
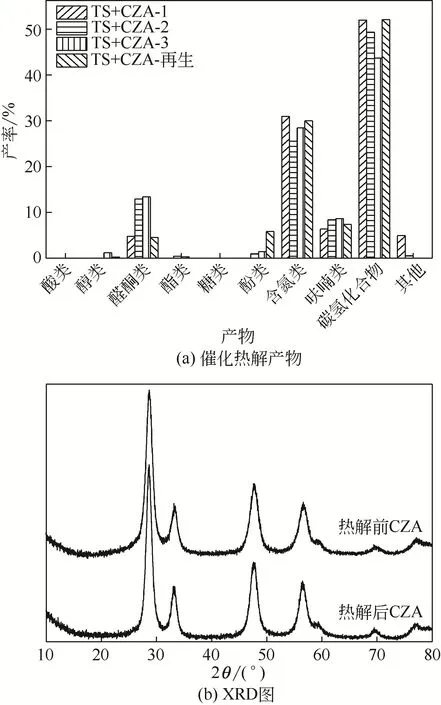
图8 CZA重复使用和再生后得到的催化热解产物及重复性测试前后的XRD谱图
图9 所示为催化剂使用一次后的TG-DTG 曲线。由图9(a)可得,催化剂的积炭量:CeO2>CZ>CZA由图9(b)可知,使用后催化剂DTG存在两个明显的失重峰,峰值温度在150℃和350℃左右,分别代表催化剂表面吸附的小分子生物油和纤维状结构焦炭。由此得出,CZ 及CZA 具有一定的抗积炭作用,主要原因可能是CeO2具有微孔结构,催化热解过程中很容易被大分子化合物阻塞,在催化剂表面聚合积炭[38]。另外,CeO2强酸活性位点较多,也容易使得催化剂表面结焦积炭。而改性后的CZ 和CZA催化剂不具备微孔结构,且催化剂表面强酸性活性位点减少,可以抑制积炭的产生。另一方面,CZ 和CZA 催化剂中CeO2占主要组分,骨架中的金属Ce 具备优良的储放氧能力,而Zr 金属的加入更加提高了催化剂的储放氧能力,催化剂表面具有更多的活性氧含量以及更快的氧迁移能力,与催化剂表面的碳化合物发生反应生成相应的气体物质,抑制了积炭效应[19]。
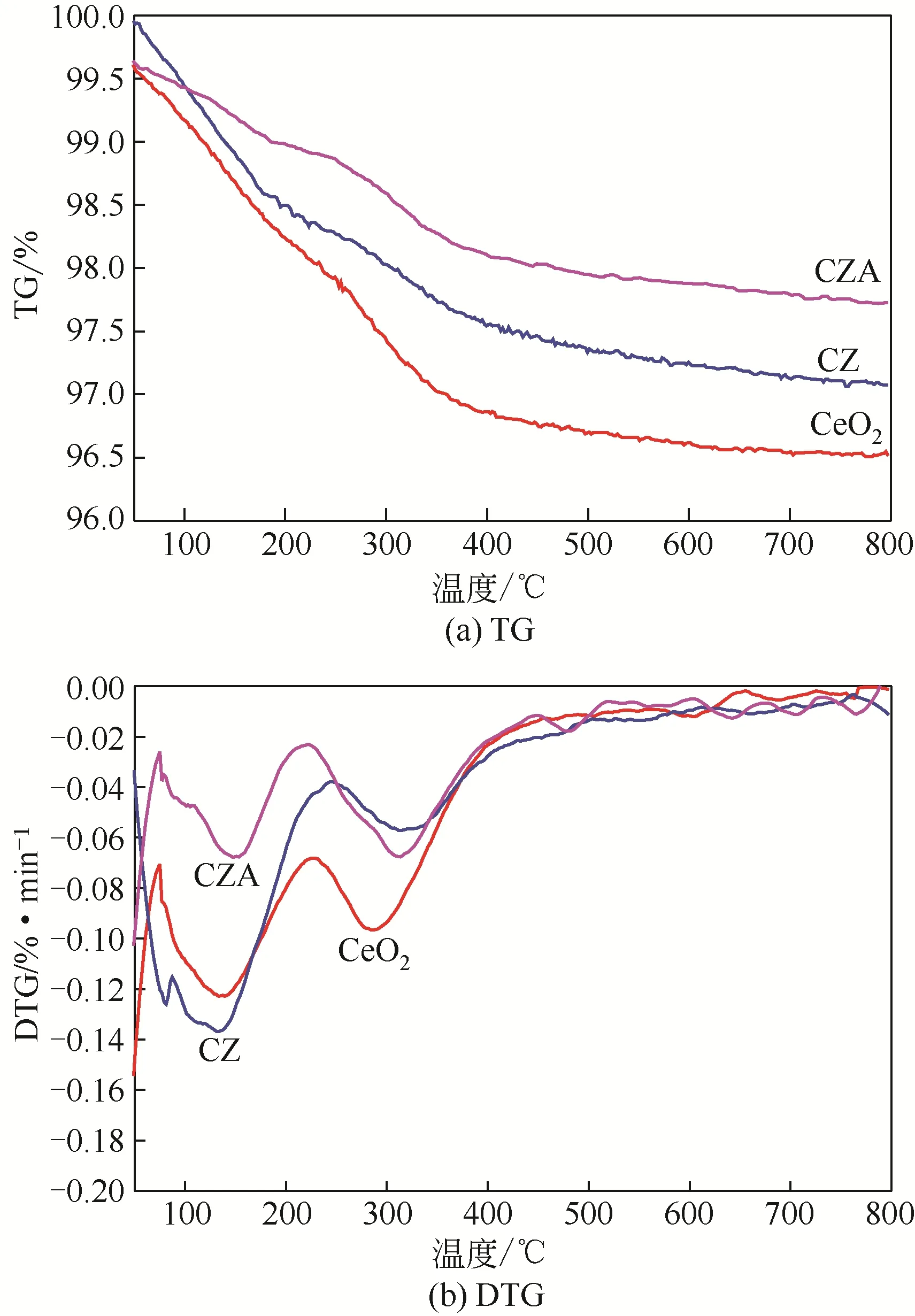
图9 催化剂使用后的TG和DTG曲线
3 结论
(1)通过共沉淀法合成的CeO2、CZ 和CZA 催化剂均属于介孔催化剂且介孔孔道较为规则。XRD结果表明CeO2为立方晶相,Zr、Al 原子的加入并没有破坏CeO2的晶型结构。NH3-TPD 结果表明,弱酸量:CZA>CZ>CeO2,CeO2的强酸量较大,CZ和CZA强酸含量降低。
(2)烟梗慢速热解产物中含氮类含量最高,达到35.2%,其次为碳氢化合物(31.17%)、酚类(10.47%)、呋喃类化合物(8.71%)和酯类化合物(7.48%)。催化剂均抑制了酯类、含氮类和酚类的形成,CeO2和CZA 抑制了醇类和呋喃类生成。三种催化剂均促进了碳氢化合物的生成,主要为芳香烃。其中,甲苯在CZA作用下达到最大值21.01%,乙苯在CeO2作用下达到最大值8.64%,二甲苯在CeO2和CZ参与下,含量分别达到9.99%和10.04%。
(3)CZA催化剂使用次数的增加导致烟梗催化热解产物中含氧化合物含量有所增加,碳氢化合物的相对含量有所降低。CZA催化剂具有良好的热稳定性以及优秀的抗积炭能力。
猜你喜欢
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
石油炼制与化工(2020年7期)2020-01-05
汽车维护与修理(2018年7期)2018-10-13
理科考试研究·高中(2014年11期)2014-11-26
燃气轮机技术(2014年4期)2014-04-16
科技传播(2010年12期)2010-04-17
数理化学习·高一二版(2009年5期)2009-07-31
数理化学习·高一二版(2009年2期)2009-03-30
