
聚氨酯硬段堆叠结构的再发现:类结晶行为
2020-06-22 03:59孔正阳应邬彬尹静波张若愚
北京化工大学学报(自然科学版) 2020年2期
孔正阳 应邬彬 胡 晗 王 凯 尹静波 张若愚*
(1.上海大学 材料科学与工程学院, 上海 200444; 2.中国科学院宁波材料技术与工程研究所, 宁波 315201)
引 言
热塑性聚氨酯弹性体是一类用途广泛的高分子材料,具有耐磨、耐热、耐腐蚀的优点以及较好的形变回复能力,它的这种优异性能与其化学结构和物理形貌有着紧密的联系[1]。热塑性聚氨酯是一种嵌段线性聚合物,通常由二异氰酸酯和低分子二醇组成的硬段相与寡聚物二醇组成的软段相构成。Bonart等[2-3]最先使用“硬段”和“软段”来描述聚氨酯结构,并用小角X射线衍射技术证实了硬相区的存在。Cooper等[4]首先提出聚氨酯弹性体的微相分离是由硬段相和软段相的热力学不相容性造成。由于氨基甲酸酯基的强极性和氢键作用,硬段分子聚集结合在一起形成硬相区,并成为物理交联点,而软段相作为基质提供弹性[5-6]。Schneider等[7]提出了一个更符合事实的微相分离结构模型,即微相分离是不完全的,除了软相区和硬链段堆叠形成的硬相区外还存在软、硬段混合的相区。除了氢键和微相分离造成的复杂形貌,结晶也有可能会对最终的结构造成影响[8]。因此,很多研究者也用差示扫描量热法(DSC)对聚氨酯进行结构研究,如Koberstein等[9]与Ryan等[10]认为在DSC升温曲线中的3个吸热峰分别为硬段微晶的熔融峰、微相分离转变温度峰以及与退火相关的峰。然而到目前为止,关于聚氨酯中的精细结构,依然没有清晰的物理图像。以往描述聚氨酯微相分离的平均场和涨落理论都缺乏对聚氨酯硬相区结构的详细描述。鉴于氨酯硬相区的精细结构往往是在一定热历史中形成的[11],因此DSC可以用来研究和分析这些精细结构的演变规律。
本文在前期研究结果[12]的基础上,利用DSC升降温实验中出现的吸热及放热峰,对聚氨酯在升降温过程中的结构演变进行探讨,特别讨论了聚氨酯硬相区中硬段堆叠的规律与方式。采用3种硬段含量逐步增加的巴斯夫(BASF)工业样品1170A、1180A和1190A,以DSC为主要表征方式结合多种测试手段来研究硬相区的形成及解离的变化规律。
1 实验部分
1.1 实验原料
3种颗粒形式的聚氨酯样品1170A、1180A、1190A均购买自德国BASF公司。本文中使用的样品都经历了两种不同的热历史:第一种为热压样品(as pressed),即将3种样品1170A、1180A、1190A分别用平板硫化机在180 ℃、185 ℃、190 ℃下热压,然后在冰水中淬火;第二种为热处理样品(annealed),即在热压样品的基础上于Linkam LTS420热台135 ℃条件下退火1 h,然后用冰水对其进行淬火处理,其热历史如图1所示。
1.2 测试与表征
1.2.1核磁共振测试
使用德国Bruker 公司的AVANCE III 400MHz型核磁共振氢谱仪(1H NMR)测试样品的核磁氢谱,条件为400.23 MHz,25 ℃,溶剂为N,N-二甲基甲酰胺(DMF)。
1.2.2XRD测试
X射线衍射(XRD)测试在德国Bruker 公司的AXS D8 Advance X射线衍射仪上进行,X射线波长1.541 Å,2θ角范围5°~45°,扫描速度5(°)/min。
1.2.3红外光谱表征
使用美国Agilent公司的Cary660+620型显微红外光谱仪进行红外光谱的表征,扫描范围500~4 000 cm-1。
1.2.4差示扫描量热表征
使用德国耐驰公司的DSC 214型差示扫描量热仪进行热分析,样品质量为5~10 mg。实验中所有的温度扫描速率均为10 ℃/min。
2 结果与讨论
2.1 核磁谱图分析
图2展示了1170A、1180A及1190A 3种聚氨酯样品的1H NMR谱图。由图可知,本文使用的聚氨酯样品组成为二苯基甲烷二异氰酸酯(MDI)、聚四氢呋喃二醇(PTMG)和1,4-丁二醇(BDO)。通过图2可以定性分析每个单体的质子峰[13]:MDI中的芳族质子位于δ=7~7.5处,MDI中的亚甲基质子位于δ=3.9处,BDO中的亚甲基质子位于δ=1.7和4.1处,PTMG中的亚甲基质子位于δ=1.6和3.4处。此外,N,N-二甲基甲酰胺中的质子位于δ=2.7~2.9和8.0处。由于分布在MDI附近的PTMG的一些氢原子的化学环境与BDO中的氢原子相同,导致部分PTMG的核磁峰与BDO重合,即δ=1.7和4.1两个位置均包含BDO和PTMG的信息。因此只可以计算图2中 “4+6”与“5+7”的比例,即只能算出MDI与PTMG+BDO的质量分数比。计算结果如表1所示,1170A、1180A与1190A中MDI的质量分数分别为23.4%、27.7%和33.5%。
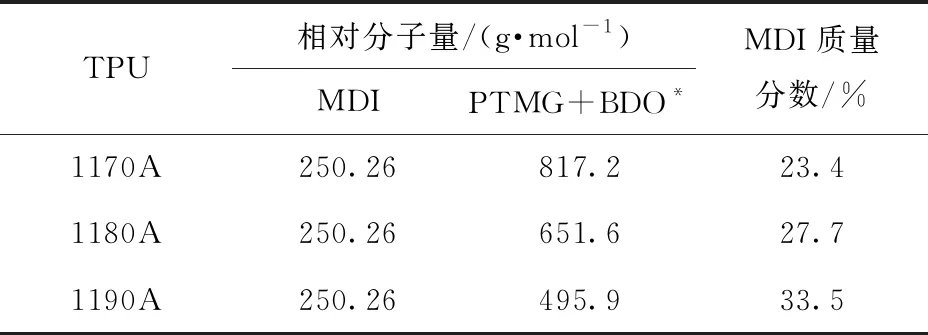
表1 利用1H NMR结果计算的聚氨酯的化学组成
*根据核磁数据计算。
2.2 聚氨酯的晶型结构
对热处理前后的1170A、1180A及1190A样品进行XRD测试,结果如图3(a)、(b)所示。显然,无论是否经过热处理,这些聚氨酯样品都没有出现明显的衍射峰,只有无定型结构的鼓包。该结果说明这3个样品无法结晶,或者结晶非常微弱[10]。以往的研究也发现,在低硬段含量的聚氨酯中硬段很难结晶[14-15]。
2.3 聚氨酯的氢键缔合程度
为了研究热历史对聚氨酯微观结构的影响,利用红外测试方法研究了压板淬冷、压板淬冷后在135 ℃或170 ℃退火并降回室温的聚氨酯的氢键缔合程度(即微相分离程度)[16-17]。如图4所示,羰基的特征伸缩振动区(1 650~1 760 cm-1)可以分解为自由羰基的伸缩振动峰(1 730 cm-1)和形成氢键羰基的伸缩振动峰(1 700 cm-1)。通过计算分峰处理后得到的两个峰的面积比可得出氢键缔合度[18-19],计算结果如表2所示。可以看出,对于拥有不同热历史的同一个样品,在室温下测得的氢键缔合度十分接近。该实验结果表明,氢键的缔合度与样品当前所处的温度密切相关,而与其之前所经历的热历史相关性较弱。此外实验结果还表明这些样品中的氢键缔合非常迅速,因为只有这样,样品才能在经历了不同的热处理过程后得到相近的氢键缔合度。这也跟本课题组之前的研究结果相符合[11]。事实上,这些实验结果与传统结果并不相同[16,20]。在传统实验中,通过时间分辨红外可以观察到聚氨酯体系随时间逐渐变化的微相分离程度。也就是表明,在传统结果中氢键缔合的过程是比较缓慢的。因此,在传统的淬火实验和降温实验中是可以观察到同一样品在室温下的不同氢键缔合度的,因为氢键缔合程度会在一定程度上被冻结。本文的实验结果拓宽了对聚氨酯的认识,也再次确认了之前我们发现的现象[12]。
同时我们也注意到,不同热历史的同一样品在室温下测得的微相分离程度略有差异。这种差异也说明即便是再快的氢键缔合速度也是有一定的时间依赖性的。聚氨酯在高温下的氢键缔合程度较低,不同的淬火或退火过程中的细微过程差异导致了微相分离程度的微小不同[21-22]。
2.4 聚氨酯的热分析结果
作为常见的热分析手段,DSC也被用来研究氢键的缔合与解离以及微相分离的发生,从而可以从热效应的角度去探索一些红外无法看到的实验现象。这里我们设计了两种不同的DSC实验,第一种实验中的样品为热压样品,而第二种实验中的样品为热处理样品。对两种DSC实验都进行3次温度扫描:第一次为60~220 ℃的升温扫描,并在220 ℃保温3 min消除热历史,第二次为220~-60 ℃的降温扫描,第三次为-60~220 ℃的二次升温扫描。这几个温度扫描的实验目的各不相同。第一次的升温扫描能很好地反映热历史对氢键缔合(硬相区)状态的影响。在经过高温消除热历史后,第二次的降温扫描能反映均相聚氨酯在降温过程中氢键的缔合行为。而第三次的升温扫描则能对降温扫描过程中形成的氢键缔合(硬相区)的解离进行分析。鉴于聚氨酯的复杂结构,这里仅讨论主要的吸热和放热峰,以抓住最主要的结构进行分析。

表2 聚氨酯红外测试的氢键缔合度
从图5(a)、(c)、(e)和表3中可以看到,这3个样品在第一次升温过程中有两个主要的吸热峰。第一个吸热峰的峰值T1都集中在53 ℃附近,而第二个吸热峰的峰值T2则随着样品中的硬段含量的增加从148.4 ℃升高到170.0 ℃。可以确定的是,T1对应的结构是在快速淬火和室温等温过程中形成的,并且3种样品中都形成了尺寸较小、结构比较杂乱和相似的硬相区,因此3种样品中的小尺寸硬相区的分解温度都比较接近。由于在淬火中仅能形成较小的硬相区,因此T2对应的是升温过程中出现的二次结构。这些二次形成的硬相区结构具有较大的尺寸和较好的规整度,对应着较高的解离温度[23]。这种现象类似于非等温快速结晶与升温过程中的重结晶。在降温过程中,3种样品都出现了一个主要放热峰T3,这是降温过程中由于硬段形成硬相区放热而形成的。此外,随着硬段含量增加,T3对应的温度也越来越高。相应地,在二次升温中,越高的T3所对应的分解温度T4也越高。这种现象类似于可结晶组分增加后,结晶与熔融温度都随之升高[24]。
另一方面,在经历了135 ℃的退火后,以上的吸热熔融温度出现了一些变化,一次升温中的主要熔融峰也变成了3个,且新出现了一个中间温度的熔融峰T5。对于T1,1170A、1180A及1190A这3个样品的变化不多。对于T2, 1170A和1180A略有升高,1190 A基本保持不变。新出现的T5转变在3种样品中的差别不是很大。此外还发现,T5对应的温度总是在退火温度(135 ℃)与T2之间。基于以上讨论可认为,在135 ℃退火中形成了一些新的硬相区结构,并且对应于T5的转变温度。而T2依然对应于在二次升温过程中小尺寸相区合并后的大尺寸结构。

表3 聚氨酯样品的DSC测试结果
为了进一步探究硬段缔合结构的形成规律,针对样品1180A进行循环变温扫描DSC实验,实验程序如图6(a)和(b)所示。在保持变温速率均为20 ℃/min的条件下,不断改变最高等温温度。图6(a)中以10 ℃为步长,从180 ℃逐步升高至250 ℃;而图6(b)中则是以10 ℃为步长,从250 ℃逐步降低至180 ℃。这里重点关注降温中硬段结构集中形成的温度范围内的放热峰峰值Tc,而不再关注升温中形成的二次结构。表4记录了各降温过程中的Tc,可以发现,在图6(c)的温度结果中,当最高温度为180 ℃和190 ℃时,Tc均为130.4 ℃,显著高于其他扫描过程中出现的Tc。这种现象表明,在最高温度为180 ℃和190 ℃时似乎并不能完全消除硬段缔合结构,并且这些残留的硬相区能够在较高温度下诱发相区的生长,出现转变温度Tc。而图6(d)的扫描结果由于是从250 ℃的高温逐步降低得到,硬相区结构已经被完全消除,Tc保持在55.8~50.6 ℃。此外需要注意的是,图6(c)中的Tc,8与图6(d)中的Tc,1的最高热历史消除温度都是250 ℃,但是图6(c)中的Tc,8略高于图6(d)中的Tc,1。也需要注意到,图6(c)和6(d)中的转变温度都在逐步降低,这说明这种硬段缔合结构在缺少足够数量的“原始硬相区”的存在下,很难进行大量生长,而硬段缔合结构即使在250 ℃的高温下,也很难在短时间内被完全清除。这种现象也跟结晶非常类似,结晶过程的第一步是成核,然后才能开始生长。从图6的结果来看,1180A样品中硬相区的出现(成核)是需要克服一定能垒的,这也跟结晶中的成核非常类似。
表4 1180A在循环升/降温中的DSC测试结果
Table 4 DSC analysis of sample 1180A in cyclic temperature scanning

升/降温程序Tc,1/℃Tc,2/℃Tc,3/℃Tc,4/℃Tc,5/℃Tc,6/℃Tc,7/℃Tc,8/℃180℃→250℃130.4130.467.967.766.364.962.560.4250℃→180℃55.854.653.253.252.451.451.450.6
以上DSC实验结果初步表明了聚氨酯吸热、放热峰的演变规律与结晶行为非常类似,这些吸、放热峰所对应的结构有着结晶中成核与生长的类似行为,它们的重整过程也与片晶的重排规律相似。同时,硬段含量越高,形成的硬段缔合结构越稳定,要破坏这些结构的温度也越高[11]。尽管在XRD的实验结果中没有显示明确的结晶信息,但是本文工作的结果暗示了这些聚氨酯样品中存在结晶结构的可能性。由于聚氨酯中氢键和微相分离的存在,聚氨酯中可能存在的结晶行为会受到这两个过程的影响,导致所形成的晶体结构可能并不完善。
3 结论
(1)在利用XRD方法对1170A、1180A及1190A这3个样品进行测试的衍射曲线中,没有发现明显的结晶结构。有意思的是,FT- IR的结果表明,不管样品经历了怎样的热处理过程,只要最终温度相同,其氢键缔合度即微相分离程度就非常接近。这样的结果表明,氢键缔合度受温度的影响非常强烈,而与热历史相关性较弱,因此也与是否会发生结晶相关性较弱。也即,微相分离与结晶的发生是相对独立的过程。
(2)DSC实验结果表明,硬段含量越高的聚氨酯越容易在高温下生成硬段缔合结构,而等温退火过程可以产生具有特征熔点的硬段缔合结构。淬火过程中生成的不同微小尺寸的硬段缔合结构都较为类似,并具有相近的熔融破坏温度。此外,在有“核”的情况下,这些硬段缔合结构的形成会更加容易。
以上结果说明,即使XRD谱图中没有明显的结晶衍射峰,这些聚氨酯材料依然显示了类似于结晶的行为。这样的结论并不支持之前一部分学者认为的DSC的吸放热峰是由氢键缔合和解离引起的结果。在未来的工作中,探索聚氨酯中与微相分离无关的潜在结晶过程是一项非常有意义的工作,一方面是因为微相分离和结晶在聚氨酯中并没有被清晰地加以区分,另一方面是结晶如何在微相区中出现尚未有过相关研究。
猜你喜欢
纺织学报(2022年3期)2022-03-28
疯狂英语·新读写(2021年5期)2021-11-23
波谱学杂志(2021年3期)2021-09-07
煤气与热力(2021年5期)2021-07-22
纺织科技进展(2021年4期)2021-07-22
纺织科技进展(2021年4期)2021-07-22
金桥(2020年10期)2020-11-26
食品与生活(2016年5期)2016-05-23
中学化学(2015年12期)2016-01-19
中学化学(2015年12期)2016-01-19
