
高效液相色谱法测定紫锥菊中7种酚酸含量及其聚类分析*
2020-06-18 01:48孙丽平齐海艳郑洪伟杜研杨春娟甘春丽
医药导报 2020年6期
孙丽平,齐海艳,郑洪伟,杜研,杨春娟,甘春丽
(1.哈尔滨医科大学药学院药物化学与天然药物化学教研室,哈尔滨 150081;2.沈阳药科大学中药学院,沈阳 110016;3.中国水利水电科学研究院,北京 100038)
紫锥菊[Echinaceapurpurea(L.) Moench]为菊科松果菊属多年生草本植物,共有8个种和数个变种,药用者主要为紫锥菊[E.purpurea(L.) Moench]、狭叶紫锥菊[Echinaceaangustifolia(DC.) Hell]和白紫锥菊[Echinaceapallia(Nutt.) Nutt],已开发为药物主要为前两种。研究表明,紫锥菊具有免疫调节、抗氧化、抗炎及抗病毒等活性,其提取物在我国保健品和药品市场有巨大的发展潜力[1-3]。紫锥菊中含有多种化学成分,如酚酸类、多糖、烷基酰胺类、倍半萜类、挥发油、生物碱类和多炔类等。主要活性成分酚酸含量较高,是《美国药典》(USP35)用于评价药材的指标性成分[4-5]。目前,我国多个地区引进紫锥菊,并且栽培成功,但有关药材质量评价和商用提取物标准的研究各异,药材质量存在较大差异[6-10]。笔者在本实验采用高效液相色谱(HPLC)法对15个产地紫锥菊提取物中7种酚酸类成分的含量进行测定,同时采用层次聚类分析法对紫锥菊提取物中酚酸成分进行分类归纳,为制定引种紫锥菊及其提取物质量标准提供参考[11-13]。
1 仪器与试药
1.1仪器 Agilent 1260色谱仪(美国安捷伦公司),Agilent 1260型二级阵列管检测器(美国安捷伦公司),HHS型电热恒温水浴锅(博讯实业有限公司医疗设备厂),RE-52AA旋转蒸发仪(上海亚荣生化仪器厂),KQ3200E超声清洗器(昆山市超声仪器有限公司),BP210S电子天平(德国Satorius公司,感量:0.1 mg)。
1.2主要试药 紫锥菊药材购于陕西、河北、湖北、湖南等15个产地,经哈尔滨医科大学药学院生药教研室鉴定为菊科松果菊属植物紫锥菊[Echinaceapurpurea(L.) Moench]。对照品:3,4-二羟基苯甲酸、绿原酸、咖啡酸、丁香酸、对香豆酸、阿魏酸、菊苣酸、香草酸、4-羟基苯甲酸均购自上海阿拉丁生化科技股份有限公司,含量均大于98.0%。无水乙醇、磷酸(分析纯,天力化学有限公司),甲醇(色谱纯,百灵威科技有限公司),纯净水(娃哈哈集团有限公司)。
2 方法与结果
2.1色谱条件 色谱柱:Inertsil C18色谱柱(250 mm × 4.6 mm,5 μm),流速:0.8 mL·min-1,检测波长:280 nm,柱温:30 ℃,进样量:10 μL,流动相:A为0.1%磷酸水溶液,B为甲醇;梯度洗脱:0~10 min,10%→20% B;>10~40 min 20% B;>40~45 min,20%→23% B;>45~50 min,2 3%→34% B;>50~63 min,34%→35% B;>63~0 min 35%→40% B;>70~75 min,40% B。在上述色谱条件下,各组分理论板数均不低于3000,样品中各待测组分与相邻峰分离度均大于1.5。
2.2溶液的制备
2.2.1对照品溶液的制备 精密称取下列对照品:3,4-二羟基苯甲酸(1)、绿原酸(2)、咖啡酸(3)、丁香酸(4)、对香豆酸(5)、阿魏酸(6)、菊苣酸(7)适量,分别用甲醇溶解并稀释成2.083,2.086,2.124,2.102,2.068,2.121,1.963 mg·mL-1的混合对照品贮备液。
2.2.2供试品溶液的制备 精密称取紫锥菊药材粉末10 g,乙醇体积分数40%,料液比1:20,50 ℃加热提取2次,每次提取2.5 h。合并提取液,滤过,减压浓缩至干,加适量甲醇溶解,转移至10 mL量瓶中,加甲醇定容摇匀,进样前用0.45 μm微孔滤膜滤过,取续滤液,即得供试品溶液。
2.3专属性实验 取3,4-二羟基苯甲酸、绿原酸、咖啡酸、丁香酸、对香豆酸、阿魏酸、菊苣酸的混合对照品溶液适量,在“2.1”项色谱条件下,进样测定,各组分理论塔板数均不低于3000,样品中各待测组分与相邻峰分离度均大于1.5,该方法专属性良好,色谱图见图1。
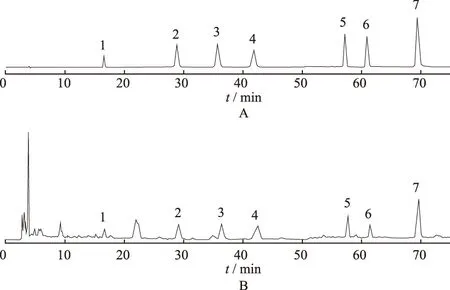
A.对照品溶液;B.紫锥菊提取物供试品溶液;1.3,4-二羟基苯甲酸;2.绿原酸;3.咖啡酸;4.丁香酸;5.对香豆酸;6.阿魏酸;7.菊苣酸。
图1 紫锥菊提取物中7种酚酸类成分HPLC色谱图
A.reference solution;B.Echinaceapurpurea extract test solution;1.3,4-dihydroxybenzoic acid;2.Chlorogenic acid;3.Caffeic acid;4.Eugenic acid;5.P-coumaric acid;6.Ferulic acid;7.Cichoric acid.
Fig.1 HPLC chromatogram of seven phenolic acids inEchinaceapurpurea extract
2.4标准曲线和线性范围 精密吸取各对照品溶液适量,置于量瓶中,用甲醇逐级稀释成3,4-二羟基苯甲酸为1.627~104.2 μg·mL-1、绿原酸为5.704~365.1 μg·mL-1、咖啡酸为3.319~212.4 μg·mL-1、丁香酸为3.284~210.2 μg·mL-1、对香豆酸为2.423~155.1 μg·mL-1、阿魏酸为3.314 ~212.1 μg·mL-1、菊苣酸为12.27~785.2 μg·mL-1的系列标准溶液,进样10 μL,测定,按“2.1”项色谱条件,记录色谱图,获取色谱峰面积,以色谱峰峰面积(Y)为纵坐标,以对照品溶液浓度(X)为横坐标绘制标准曲线,进行线性回归分析。结果见表1,各组分在其相应的浓度范围内线性关系良好(R2> 0.999 6)。
表1 紫锥菊提取物中7种酚酸类成分的标准曲线与线性范围
Tab.1 Standard curve and linear range of seven phenolic acids in Echinacea extract

化合物线性方程R2线性范围/(μg·mL-1)3,4-二羟基苯甲酸Y=8.295 5X-1.495 50.999 91.627~104.2绿原酸Y=8.787 6X+2.801 30.999 85.704~365.1咖啡酸Y=20.783X+6.612 30.999 73.319~212.4丁香酸Y=17.599X+10.6320.999 73.284~210.2对香豆酸Y=25.289X+3.639 80.999 82.423~155.1阿魏酸Y=18.645X-2.236 40.999 93.314~212.1菊苣酸Y=11.463X-126.090.999 612.270~785.2
2.5精密度实验 取同一混合对照品溶液,按“2.1”项色谱条件连续进样6次,进行测定,记录化合物1-7各对照品的峰面积,并计算各峰面积的相对标准偏差值(RSD)。结果RSD分别为2.04%,2.16%,2.06%,1.98%,2.00%,2.02%,1.27%,结果表明本实验所用仪器精密度良好。
2.6重复性实验 取同一产地(陕西)紫锥菊药材粉末6份,每份10 g,精密称定,按“2.2”项下方法制备供试品溶液,按“2.1”项下色谱条件进样分析,测定7种酚酸成分的峰面积,外标法计算浓度及其RSD值。结果化合物1-7浓度的RSD值分别为1.15%,0.86%,2.04%,0.37%,1.98%,1.60%,0.96%,结果表明本实验提取测定方法重复性良好。
2.7稳定性实验 取同一混合对照品溶液,室温放置,分别在0,2,4,6,8,12 h分别进样10 μL,按“2.1”项色谱条件进样分析,外标法计算各时间隔点7种酚酸成分的浓度,并与0 h的浓度比较,计算RE值。结果见表2,表明7种酚酸在12 h内稳定性良好。
2.8加样回收率实验 取同一产地(陕西)紫锥菊药材粉末6份,每份5 g,精密称定,加入适量化合物1-7到供试样品中,按“2.2”项下方法制备供试品溶液。按“2.1”项下色谱条件进样分析,获取色谱峰面积,外标法计算浓度,计算各酚酸成分的加样回收率及RSD值。结果显示加入7种酚酸成分样品的平均加样回收率为98.41%~101.9%,RSD<3.0%,表明本实验方法准确可靠。
2.9样品测定 取不同产地同一季节采收的紫锥菊药材各10 g,精密称定,按“2.2”项方法制备供试品溶液,按“2.1”项色谱条件进行分析,记录色谱峰面积,用外标法计算各产地紫锥菊提取物中7种酚酸成分的含量及总酚含量。结果见表3。

表2 7种酚酸稳定性实验结果
2.10不同产地紫锥菊提取物中7种酚酸类成分的聚类分析 聚类分析是一种依据研究对象(样品或指标)的特征,对其进行分类的方法,以减少研究对象的数目。本实验根据所测不同产地紫锥菊提取物中7种酚酸成分的定量分析数据,使用SPSS 22.0版统计软件处理,选用离差平方和法(Ward法),平方欧式距离(Squared Euclidean distance)作为样品的测度,进行层次聚类分析。得到不同产地紫锥菊提取物中7种酚酸类成分含量的树状聚类图谱,结果见图2。
由图2可知,样本层次聚类分析结果根据不同产地紫锥菊提取物中酚酸类成分含量的不同大体可以分为两类:样本3,7,1,2是一类,说明这类紫锥菊提取物中7种酚酸类成分含量基本相近,剩余样本为一类。剩余样本也可以继续细分:样本6,13,14为一类,样本4,8,10为一类,样本5,9,12,15,11为一类。从分析结果可以看出,不同地区的样品各自聚为一类,说明紫锥菊提取物中酚酸类成分含量显然与地域分布有关。
3 讨论
3.1色谱条件的选择 实验中进行色谱条件的优化,由于7种酚酸类成分极性相近,为了达到良好的分离度,本实验采用梯度洗脱。结合文献,考察不同流动相对样品分离效果的影响。有机相采用甲醇-水相考察0.1%磷酸水溶液、0.2%磷酸水溶液和0.5%磷酸水溶液,分别组合挑选最优流动相。经过多次调整洗脱条件,最终确定以甲醇-0.1%磷酸水溶液系统作为流动相,7种酚酸类成分得到较好地分离,且柱效较高,拖尾现象得到改善。针对酚酸类成分母核中含有苯环结构,选用DAD检测器进行测定,在280 nm波长处各组分均有较大的吸收,确定为检测波长。
3.2测定结果分析 表3数据显示不同产地紫锥菊提取物的酚酸含量参差不齐,其中陕西产地紫锥菊提取物的酚酸总含量最高,而湖南的最低,两者含量相差明显。同时紫锥菊提取物中菊苣酸含量相比于其他成分要高,其次是绿原酸和咖啡酸,说明这3种成分是紫锥菊提取物中主要的酚酸类成分。总的来说,紫锥菊提取物中总酚酸及指标性成分菊苣酸、绿原酸和咖啡酸的含量因产地不同而不同,初步推测是由于药材的生长环境,采摘、干燥和贮存等环节中存在差异造成的。《美国药典》中采用标准提取物定性,对绿原酸对照品及一测多评校正因子进行定量,加和,计算总酚酸含量,三者总含量不少于4%。本研究采用直接测定法测定7种酚酸类成分的含量,15个产地酚酸总含量均未达到4%,最高仅约为1%。初步分析其原因可能有:一是引种地与原产地的自然条件存在差异,可能造成酚酸类成分含量变化;二是采摘时期、贮存场所、干燥方式等药材处理不规范造成酚酸类成分的降解;三是在紫锥菊引种驯化过程中生物学因素,如遗传特性等改变也会造成体内次级代谢产物的变化。

表3 不同产地紫锥菊提取物中7种酚酸类成分的含量测定结果

1.安徽;2.江苏;3.湖北;4.云南;5.河南;6.甘肃;7.湖南;8.四川;9.山西;10.广西;11.北京;12.河北;13.新疆;14.陕西;15.山东。
图2 15个产地紫锥菊提取物中7种酚酸类成分含量聚类分析树状图
1.Anhui;2.Jiangsu;3.Hubei;4.Yunnan;5.Henan;6.Gansu;7.Hunan;8.Sichuan;9.Shanxi;10.Guangxi;11.Beijing;12.Hebei;13.Xinjiang;14.Shaanxi;15.Shandong.
Fig.2 Tree diagram of clustering analysis on the contents of seven phenolic acids in Echinacea extracts from 15 habitats
因不同产地紫锥菊提取物中酚酸类成分的含量数据比较分散,对其相似程度进行直观比较,很难做出相应的结论。为获得准确的结论,本研究利用化学识别模式中的聚类分析方法对分散的数据标准化处理,按紫锥菊药材产地进行分类归纳。由图2可知,紫锥菊提取物中酚酸类成分含量的聚类显示出一定的地域相关性,根据样本层次聚类分析结果大体上可以分为两类:样本湖北、湖南、安徽、江苏是一类,剩余样本为一类,表明相邻省份提取物含量相近。聚类分析结果可为紫锥菊药材质量的一致性和合理种植提供实验依据。
猜你喜欢
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
天然产物研究与开发(2019年1期)2019-03-01
中成药(2018年11期)2018-11-24
中成药(2018年9期)2018-10-09
天然产物研究与开发(2018年7期)2018-08-21
中成药(2018年7期)2018-08-04
中成药(2018年2期)2018-05-09
中成药(2017年10期)2017-11-16
中国畜牧兽医文摘(2016年3期)2016-01-31
爱你(2015年6期)2015-04-20
