
以中枢性尿崩症为首发表现的肝豆状核变性1例报告
2020-06-16 09:15:28赵茂,刘蕊,胡晓
临床肝胆病杂志 2020年5期
赵 茂, 刘 蕊, 胡 晓
1 遵义医科大学及其附属医院, 贵州 遵义 563003; 2 贵州省人民医院, 贵阳 550001
肝豆状核变性(hepatolenticular degeneration,HLD),又称Wilson病,是一种常染色体隐性遗传的铜代谢障碍性疾病[1],该病在世界范围内的发病率为1/3万~1/10万[2],是至今少数早期诊断及正确治疗可以得到较好疗效的遗传代谢性疾病之一。其主要发病机制为13号染色体上ATP7B基因纯合或复合杂合突变,导致其编码产物ATP7B的功能缺陷,引起血清铜蓝蛋白合成减少及胆道排铜障碍,大量游离铜离子沉积于肝、脑、角膜等处造成相应器官损伤[1,3]。由于铜在不同部位的异常沉积,临床表现多样,最常见为肝损伤、锥体外系症状、角膜色素环[4]。以中枢性尿崩症为首发症状的HLD罕见,容易延误诊断,现将贵州省人民医院收治的1例报道如下。
1 病例资料
患者男性,19岁,因“烦渴、多饮、多尿1个月余”于2017年10月8日就诊贵州省人民医院内分泌科。患者在1月余前感冒后出现烦渴、多饮、多尿。饮水量约10 L/d,尿量与饮水量相当,期间因口渴未能及时饮水出现畏寒、发热,体温最高达38.5 ℃,伴乏力。上述症状持续无好转。既往史、家族史无特殊。入院查体:生命体征平稳,体型消瘦,面部可见蜘蛛痣1枚,双下肢皮肤可见色素沉着。腹软,无压痛及反跳痛。肝肋下未触及,脾左锁骨中线和肋缘交点下6 cm可扪及。
入院后完善相关检查,血常规:WBC 2.66×109/L,RBC 4.22×1012/L,Hb 118 g/L,PLT 54×109/L;肝功能:AST 102 U/L,GGT 123 U/L,ALP 227 U/L,总蛋白47.5 g/L,白蛋白22 g/L,球蛋白25.5 g/L,白球比值0.86。尿渗透压49 mosm·kg-1·H2O-1,尿比重1.005,行禁水加压素试验提示中枢性完全性尿崩症。腹部B超示:肝硬化并脾脏肿大、胆囊壁水肿;肝硬度示:脂肪衰减值171 dB/m, 肝硬度值12.2 kPa。上腹部增强CT示:肝硬化征象,脾大、腹水、门静脉高压、侧支循环开放。请感染科会诊后查:血清铜蓝蛋白13.3 mg/dl,血清铜601.7 μg/L,24 h尿铜417.9 μg。头颅MRI平扫示:脑白质散在缺血灶(Fazekas 1级),磁敏感加权成像未见异常。垂体MRI平扫及增强示:垂体后叶短T1高信号消失(图1)。请眼科会诊后行双眼眼前节照相,双眼角膜缘可见K-F环(图2)。
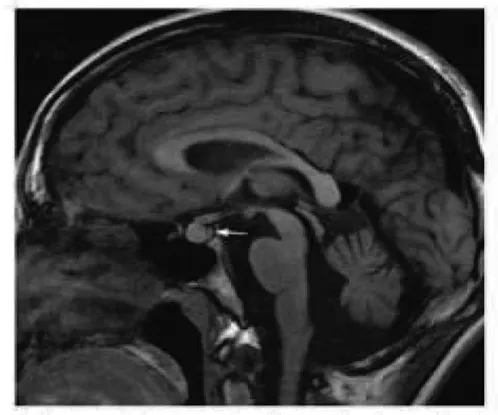
注:垂体后叶短T1高信号消失(箭头所示)。
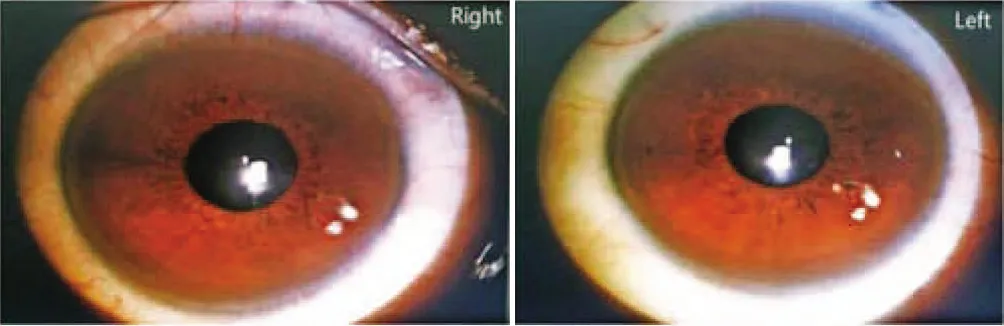
注:双眼角膜缘可见K-F环。
根据患者症状体征及实验室等检查结果,考虑HLD可能性大,但患者除肝损伤外,尿崩症症状突出,临床表现不典型,为进一步确诊,在患者及其家属的知情同意下,对患者及其家属进行基因检测。采用Sanger测序法,对ATP7B基因21个外显子及侧翼序列进行突变分析,结果发现,该患者ATP7B基因存在两个杂合致病突变:2299dupC(Met769Hisfs*26),2827G>A(Gly 943Ser)。其母亲基因检测为2299dupC单位点突变,单纯杂合子。其父亲基因检测为2827G>A 单位点突变,单纯杂合子。父母均无症状,尿铜正常。
最终诊断为HLD合并中枢性尿崩症,予低铜饮食、青霉胺联合硫酸锌合剂驱铜、补锌、醋酸去氨加压素口服等对症治疗。经治疗后,复查肝功能:AST 46 U/L,ALP 210 U/L,GGT 85 U/L;复查24 h尿铜:189 μg,同治疗前相比呈下降趋势,驱铜治疗有效,患者临床症状好转,入院14 d后带药出院。随访1年半,患者多尿症状明显减轻,但仍反复出现腹水等肝硬化症状,继续治疗中。
2 讨论
HLD是一种铜代谢障碍所致疾病,为常染色体单基因隐性遗传病。HLD患者多数以肝病和神经精神症状起病,少数以肾损害、骨关节损害及血液系统症状等起病[5]。由于该病常涉及多个器官系统,临床表现复杂多样,且病程进展不同,早期容易误诊及漏诊[6-7]。本例患者以尿崩症起病,虽有肝脏受损表现,但尿崩症症状突出,禁水加压素实验及垂体MRI检查,提示有中枢性尿崩症,是否与HLD相关,值得进一步探讨。
中枢性尿崩症是一种较为少见的内分泌系统临床综合征,主要以多尿、烦渴、多饮、低比重尿及低渗透压尿为特征。其发病机制主要为下丘脑神经垂体病变导致抗利尿激素分泌不足。本例患者以多尿、烦渴、多饮起病,垂体MRI及禁水加压素实验有中枢性尿崩症特征,但既往无颅脑外伤及手术史,而且查头颅CT未见异常钙沉积高信号,头颅MRI未见异常铁沉积及占位病变,垂体MRI未见炎症及肿瘤病变,可排除单纯性尿崩症、颅脑外伤及手术、垂体肿瘤及炎症、异常钙或铁沉积导致垂体病变,推测中枢性尿崩症为铜异常沉积所致。临床进一步证实血清铜蓝蛋白明显下降、角膜K-F环,且患者存在无法解释的肝硬化,说明体内存在铜代谢异常并累及多系统。最终经基因检测证实为HLD。因此,考虑患者出现中枢性尿崩症等内分泌紊乱是由HLD导致的。
目前尚无临床案例报道,明确提及HLD表现为尿崩症者。但既往临床报道中有HLD患者出现垂体功能减退症[8]、下丘脑-垂体-性腺轴功能损害[9-10]、多囊卵巢综合征[11]等内分泌系统受累表现;与本例患者类似,因此推测患者不考虑单纯性尿崩症,而是HLD导致继发性中枢性尿崩症。由HLD导致中枢性尿崩症发生的可能机制为铜代谢异常致垂体内分泌紊乱。由于铜离子在下丘脑垂体的沉积,破坏下丘脑、垂体细胞及亚细胞的结构和功能,使下丘脑-垂体功能缺陷或受损,导致垂体分泌抗利尿激素功能下降,产生中枢性尿崩症。
HLD患者的影像学特点为病变范围分布广泛,而且由于疾病的不同发展时期和MRI系统的特性使病变表现具有多样性[12]。既往研究[4]已表明,HLD患者头颅MRI可发现豆状核(尤其壳核)、尾状核、中脑、脑桥、丘脑、小脑及额叶皮质Tl加权像低信号和T2加权像高信号;近期研究者[13-14]发现,应用磁共振弥散张量成像、静息态功能磁共振、磁敏感成像等影像学检查可检测HLD患者脑部丘脑等核团存在结构性损伤及功能活动水平的下降。本例患者因多饮、多尿就诊,头颅MRI提示脑白质散在缺血灶(Fazekas 1级),磁敏感加权成像检查未见明显异常,非典型HLD影像学改变。为进一步确诊,患者完善血清铜蓝蛋白、角膜K-F环及ATP7B基因检测,结果均支持HLD诊断。因此,对临床疑似HLD患者,尤其头颅MRI等影像学表现不典型患者,应尽早完善铜代谢生物化学、K-F环等特异性检查,并结合基因检测进行综合判断。
基因诊断是目前HLD早期确诊的最有效方法[1],尤其对临床症状、生化及影像学特点不典型患者有重要意义。国内外指南[4,15]也一致推荐,对任何临床及生化检查难以确定的疑似HLD均应进行基因检测。ATP7B基因是目前已知唯一的HLD致病基因,但其突变位点及类型复杂,而且该基因突变具有种族差异[1],欧美HLD患者以p.His1069Gln突变最多见[16]。在我国最常见的突变类型为p.Arg778Leu,其次为p.Pro992Leu,相关研究[17]也表明我国HLD患者基因突变以少数几个热点突变和广泛罕见突变为特征。HLD患者的基因水平确诊需满足ATP7B基因纯合致病突变或复合杂合致病突变,单一杂合突变不能确诊[18],一项对ATP7B基因突变位点的研究[19]也发现,复合突变者常比单一突变者更早发病。结合本例HLD患者,ATP7B 基因测序为p.Met769Hisfs*26和p.Gly 943Ser两个位点的突变,为复合基因突变型,其父母均发生一种杂合突变,表型正常,故可确诊患者因遗传父母双方的杂合突变而发生HLD。
综上所述,对于中枢性尿崩症患者,合并不明原因的肝脏病变需警惕HLD,临床医生应仔细询问病史,同时要平行检查血清铜蓝蛋白、血铜、24 h尿铜、角膜K-F环、ATP7B基因等多项指标,请感染科、眼科等相关科室会诊,以降低误诊率和漏诊率,使患者得到早诊断、早期干预治疗,最终改善疾病预后。
猜你喜欢
现代实用医学(2022年10期)2022-12-08 05:48:32
家庭医药(2021年15期)2021-12-01 22:46:06
家庭医药(2021年8期)2021-07-28 23:18:42
种子(2021年3期)2021-04-12 01:42:22
中国实用医药(2016年19期)2016-08-05 22:41:35
外语教学理论与实践(2016年1期)2016-06-11 05:51:48
中国继续医学教育(2015年3期)2016-01-06 01:36:40
医学研究杂志(2015年9期)2015-07-01 17:28:15
华东理工大学学报(自然科学版)(2014年1期)2014-02-27 13:48:29
中国医学科学院学报(2013年2期)2013-03-11 20:25:43
