
SCCD的发病机理及其致病基因UBIAD1的功能研究进展
2020-06-08 00:25苏振宏黄玉迪张军林陶俊峰解举民
国际眼科杂志 2020年6期
苏振宏,黄玉迪,张军林,陶俊峰,袁 超,解举民
0引言
施奈德结晶状角膜营养不良(Schnyder crystalline corneal dystrophy,SCCD,MIM 121800)是一种稀有的常染色体显性遗传病,患者双眼角膜随年龄增长逐渐混浊化,导致视力衰退,最终丧失视力,SCCD在男女中患病几率均等,发病原因为眼角膜中胆固醇和磷脂的异常积累[1]。SCCD的分子基础为1号染色体短臂3区6带的UBIAD1基因突变[2-3],该基因编码一个含有338个氨基酸的蛋白质,分子量为36.8kDa,含有一个异戊烯转移酶结构域(aa:58-333)。UBIAD1基因又被称为移行上皮反应基因(transitional epithelial response gene,TERE1)[4-5]。现已证实,UBIAD1突变可以导致SCCD的发生[2-3],但是致病的分子机制尚不清楚。日本学者证明UBIAD1是一种存在于人体内的4-甲基萘醌合成酶[6],参与细胞中脂质代谢。Fredericks等[7]将UBIAD1(TERE1)导入HEK293细胞和膀胱癌细胞,发现细胞中胆固醇的积累量增长了30%,而且在人膀胱移行细胞癌J82细胞中,UBIAD1的表达量下调了1/3,证实UBIAD1与细胞内的胆固醇代谢密切相关。SCCD的临床表现为角膜中胆固醇与磷脂的异常积累[1]。UBIAD1基因突变后导致了SCCD的发生,且SCCD是一种连续性发展的疾病,随着年龄的增长,脂质在角膜处积累逐渐增多,导致角膜混浊化,视力衰退。本文旨在系统、全面地综述SCCD致病基因UBIAD1的突变与疾病发生的关系,并通过UBIAD1基因的功能分析其突变后导致SCCD发生的分子机制。
表1 UBIAD1基因突变导致SCCD的位点
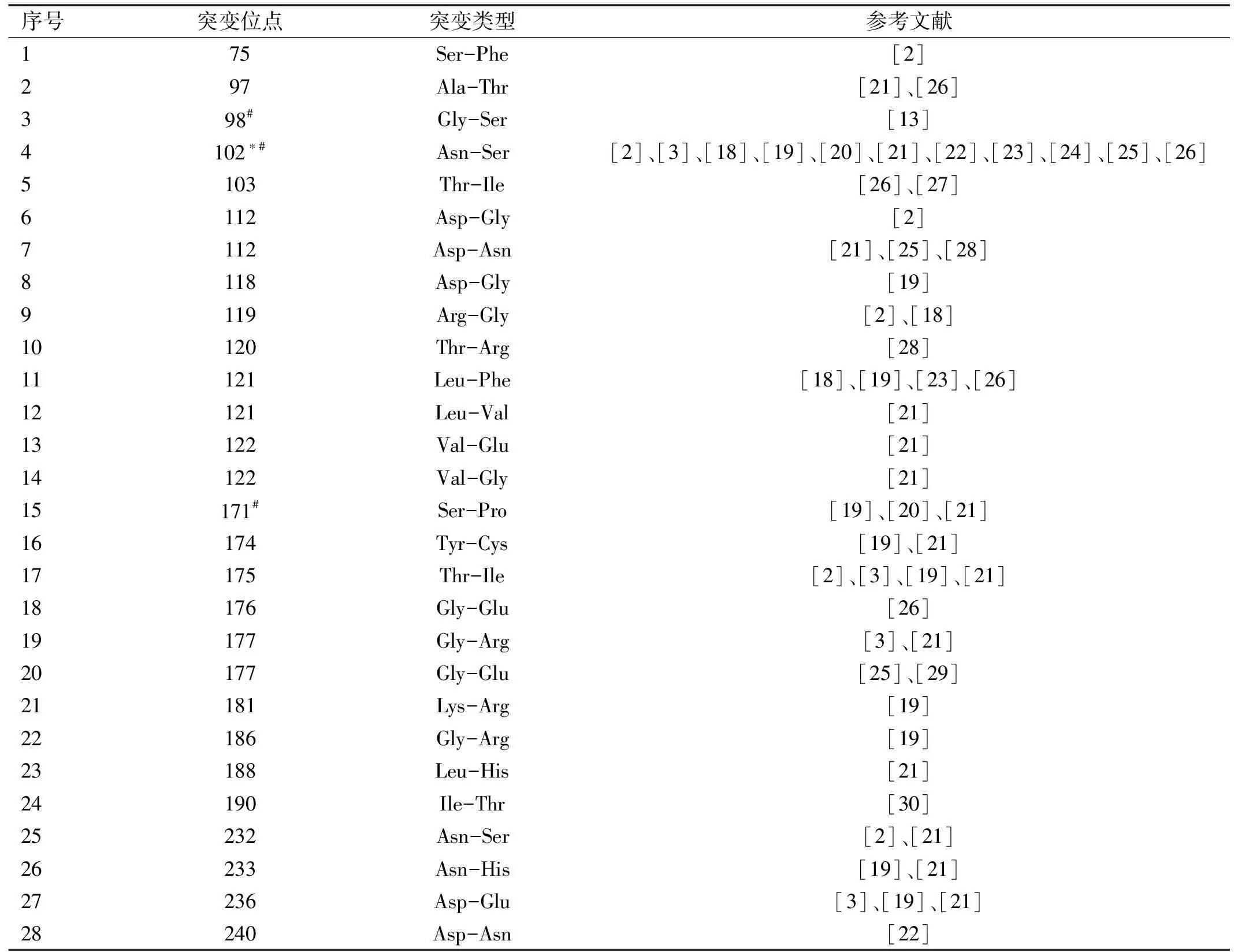
序号突变位点突变类型参考文献175Ser-Phe[2]297Ala-Thr[21]、[26]398#Gly-Ser[13]4102∗#Asn-Ser[2]、[3]、[18]、[19]、[20]、[21]、[22]、[23]、[24]、[25]、[26]5103Thr-Ile[26]、[27]6112Asp-Gly[2]7112Asp-Asn[21]、[25]、[28]8118Asp-Gly[19]9119Arg-Gly[2]、[18]10120Thr-Arg[28]11121Leu-Phe[18]、[19]、[23]、[26]12121Leu-Val[21]13122Val-Glu[21]14122Val-Gly[21]15171#Ser-Pro[19]、[20]、[21]16174Tyr-Cys[19]、[21]17175Thr-Ile[2]、[3]、[19]、[21]18176Gly-Glu[26]19177Gly-Arg[3]、[21]20177Gly-Glu[25]、[29]21181Lys-Arg[19]22186Gly-Arg[19]23188Leu-His[21]24190Ile-Thr[30]25232Asn-Ser[2]、[21]26233Asn-His[19]、[21]27236Asp-Glu[3]、[19]、[21]28240Asp-Asn[22]
注:#:中国人群中发现的UBIAD1突变; *:UBIAD1突变热点。
1 SCCD的临床表现
SCCD是一种稀有的常染色体显性遗传病,其发病特点为连续性双眼角膜混浊化,原因为胆固醇、磷脂等在角膜中异常积累。大部分SCCD患者血脂、脂蛋白和胆固醇含量异常,但是角膜逐渐混浊化和结晶化与血脂并没有直接关系[8]。临床检测发现约54% SCCD患者角膜中会出现结晶状沉淀,其余46% SCCD患者并没有检测到角膜结晶状沉淀,所以国际命名委员会应Weiss等眼科学家的要求将SCCD重新命名为SCD(Schnyder corneal dystrophy,SCD)[9-11](为避免混淆,本文统一描述为SCCD)。
SCCD的临床表现根据发病年龄可以划分为3个阶段[8]:(1)26岁及以下,表现为在角膜基质上皮下中央位置出现结晶状沉淀;(2)27~39岁,表现为结晶状沉积不断积累,并出现薄雾化;(3)40岁及以上,表现为随着年龄的增长,结晶化程度不断加重,导致整个角膜混浊化。根据角膜中结晶状沉淀的积累推知该病随年龄的增长呈连续性发展,导致视力逐渐缺失,最终失明。约4% SCCD患者会伴有膝外翻或叉型腿等表型[1,8,12-13]。SCCD患者主要通过穿透性角膜移植(penetrating keratoplasty,PKP)和光治疗性角膜切削术(phototherapeutic keratectomy,PTK)两种方法治疗,恢复部分视力[8]。通过化学方法检测SCCD患者手术移除的眼角膜中胆固醇的含量发现,胆固醇含量增加了10倍,磷脂含量增加了5倍,高密度脂蛋白(high density lipoprotein,HDL)中的载脂蛋白异常积累[14]。
2 SCCD发生的分子基础
SCCD最早由法国眼科医师Van Went和Wibaut于1924年报道,他们在1个家族中发现8例患者双眼角膜均出现混浊化[15]。1927年、1929年、1939年瑞士眼科医生Schnyder先后阐明本病的临床表现与遗传特性,故此病被称为施奈德结晶状角膜营养不良[16-17]。直至2007年,该疾病的分子基础才被Orr等[2]、Weiss等[3]、Yellore等[18]研究揭示,证明位于1号染色体短臂3区6带的UBIAD1基因突变可导致胆固醇、磷脂等脂质在角膜处异常积累。随后,世界各地的科学家及眼科学家,不断发现和报道新的SCCD病例。通过序列分析得到多个UBIAD1点突变,证明该基因突变可以导致SCCD的发生,进一步阐明了SCCD发病的分子基础。
截止目前,通过文献检索共发现28个可以引起SCCD发生的UBIAD1点突变,其中UBIAD1第102位天冬氨酰突变是所有点突变中的热点,现已在多个SCCD家系中发现该突变[2,3,18-25,26]。国内学者报道的SCCD家系中也发现了102位氨基酸突变,同时还发现了98位和171位氨基酸突变[13,19-21]。我们总结了从2007年SCCD分子基础被揭示以来至2019年发现的导致SCCD发生的全部UBIAD1点突变[27-30],见表1。SCCD在欧美国家的发病率高于其他国家和地区,分析可能与UBIAD1基因的易感性有关。Dong等[31]运用CRISPR-Cas9技术构建了SCCD小鼠模型,将小鼠UBIAD1基因的第100位氨基酸引入突变,由天冬氨酰突变为丝氨酸,该位置对应于人第102位热点突变,为SCCD的治疗与发病机理的研究提供了动物模型。尽管SCCD的分子基础已经被证实,但其具体发病机制尚不清楚。
3 SCCD致病基因UBIAD1的功能
UBIAD1基因于2001年被发现,在人体的多种组织中表达,但是在侵入性移行细胞癌中该基因表达量降低或不表达。将该基因转入移行细胞癌TCC细胞中,发现细胞的增殖抑制率达80%~90%[4],但是该基因的具体功能未知。McGarvey等[5]证明,25%的膀胱肿瘤中未检测到UBIAD1表达,将UBIAD1基因转入LNCaP和PC-3两种膀胱癌细胞系中,细胞生长抑制率达80%,表明UBIAD1可以明显抑制膀胱癌细胞增殖,推测该基因是潜在的抑癌基因。2010年,Nakagawa等[6]证明UBIAD1是人体中的MK-4生物合成酶,参与细胞中胆固醇代谢,UBIAD1基因的功能研究随之成为热点。有研究通过基因芯片分析发现,在1/3的膀胱癌样本中,UBIAD1的表达量减少。在裸鼠膀胱癌模型中表达UBIAD1,可以抑制肿瘤的发生发展。UBIAD1突变可影响其与APOE蛋白结合,导致细胞内胆固醇水平异常,从而提高癌细胞中的胆固醇含量。若移行细胞癌中缺失UBIAD1表达,MK-4介导的胆固醇平衡则被打破,癌细胞持续增殖[7]。
UBIAD1是一种异戊烯转移酶,在维生素K2和辅酶10(CoQ10)的生物合成中起到重要作用。UBIAD1亚细胞定位在线粒体、内质网和高尔基体上。研究证明,UBIAD1与线粒体中TBL2蛋白存在相互作用,异源表达UBIAD1可以提高线粒体跨膜电位,氧化压力和NO产量,激活SXR靶标。UBIAD1-TBL2复合物在氧化压力、硝化压力、脂代谢和SXR信号通路中具有重要作用[32]。另有研究报道,在肾透明细胞癌中表达UBIAD1,可以增加线粒体耗氧量、氢产量、氧化压力和NO产量,降低胆固醇含量[33]。Hegarty等[34]研究发现,在多器官的脊椎动物中UBIAD1对血管内皮细胞的生存和发育至关重要,其原因可能是UBIAD1介导维生素K2的合成,而且该基因还具有调节心脏功能的作用,斑马鱼UBIAD1同源基因reddish/reh突变后,斑马鱼会出现心脏水肿、颅内出血、血管退化等表现。Mugoni等[35]利用斑马鱼研究发现,UBIAD1直系同源基因barolo/bar突变的斑马鱼心血管系统发育失败,原因为氧化压力以及ROS介导的细胞损伤。UBIAD1是一个定位于高尔基体的异戊烯转移酶而非定位于线粒体,在高尔基体上合成CoQ10,UBIAD1缺失可导致细胞基质中抵御氧化压力的CoQ10减少,血管壁细胞中ROS介导的脂质过氧化。在心血管中抑制eNOS可以阻止UBIAD1依赖的氧化损伤,表明UBIAD1可通过调控CoQ10的合成调节eNOS的活性,在NO信号通路中起重要作用[36]。
敲除小鼠UBIAD1基因,小鼠胚胎成活7.5d后发育停止。UBIAD1-/-胚胎干细胞中不能合成维生素K2,但是可以合成CoQ9,与野生型胚胎干细胞相似。UBIAD1+/-小鼠可以正常发育和交配,其组织中维生素K2的合成量与含量约是正常小鼠组织的1/2,但是CoQ9的含量与野生型小鼠相比大致相等。UBIAD1-/-小鼠胚胎无法存活,但是给UBIAD1+/-孕母鼠补充MK-4或者CoQ10可以延长胚胎的生存时间。UBIAD1在维生素K2的合成中起作用,但与CoQ9的合成不相关[37]。为了解决这一难题,Nakagawa团队利用5a时间构建了他莫昔芬诱导的UBIAD1基因敲除鼠,该小鼠在喂食他莫昔芬后,生存期延长至60d,小鼠发育成熟,他们用此模型来研究UBIAD1基因敲除后小鼠的变化,解剖发现小鼠的胰脏体积变小,胰脏腺泡细胞变成液泡细胞,鉴定后发现,该液泡细胞为脂肪细胞。UBIAD1基因缺失后,胰脏腺泡细胞氧化压力和自噬增强,导致细胞出现凋亡。这一结果表明UBIAD1基因对胰脏腺泡细胞的生存至关重要,而且对细胞内脂质的代谢具有重要作用[38]。
Hirota等[39]借助生信分析方法及生化检测手段将UBIAD1蛋白的4个保守结构域逐一进行分析发现,保守域Ⅰ(101~133)参与了底物的结合;保守域Ⅱ(140~150)是氧化还原反应结构域,其中包含CxxC结构域;保守域Ⅲ(169~184)铰链区是重要的催化位点中心;保守域Ⅳ(240~253)是Mg2+和异戊二烯基结合位点。该研究为进一步揭示细胞中UBIAD1蛋白的工作机制奠定了基础。UbiA超家族蛋白是一种跨膜的异戊烯转移酶,控制辅酶Q、维生素K2、质体醌、亚铁血红素、叶绿素、维生素E和结构脂质生物合成的关键步骤,上述脂溶性化合物常作为电子或质子供体参与细胞呼吸、光合作用,也作为抗氧化剂保护细胞免受氧化损伤[40]。UBIAD1蛋白是UbiA超家族蛋白成员之一,其在慢性肾脏疾病和心血管疾病中也起到重要作用,推测其通过调节细胞中MK-4和CoQ10的生物合成调控细胞中胆固醇水平,进而影响肾脏细胞和心血管细胞的钙化、凋亡和增殖过程[41-42]。Huang等[43]发现在N2A 细胞中UBIAD1通过调控PI3K/AKT信号通路保护因为缺氧和低糖造成的亚细胞器损伤。
UBIAD1突变可引起脂质在眼角膜中的异常积累,导致SCCD的发生[2-3]。研究发现在哺乳动物细胞中固醇类物质可以刺激UBIAD1与HMG CoA还原酶(3-羟基-3-甲基戊二酰辅酶A)结合,牻牛儿基牻牛儿基焦磷酸(GGpp)存在时,GGpp可与UBIAD1蛋白的催化中心结合,UBIAD1-HMG CoA还原酶复合物解离,之后UBIAD1被转运到高尔基体,HMG CoA还原酶破膜进入细胞质中被蛋白酶体降解,具体分子机制见图1[44-45]。当UBIAD1出现点突变以后,其构象发生改变,如最常见的N102S点突变,UBIAD1突变体不能与GGpp结合,导致UBIAD1-HMG CoA还原酶复合物不发生解离,一直存在于内质网膜[44-45]。HMG CoA还原酶是胆固醇和非甾醇类异戊二烯生物合成中的限速酶,HMG CoA还原酶的正常表达是细胞胆固醇和非甾醇类异戊二烯物质合成的基础。当HMG CoA还原酶不能被及时降解时,可导致胆固醇和非甾醇类异戊二烯物质在细胞内积累[46-48]。因此我们推测,SCCD的致病分子机制为UBIAD1基因突变后,导致UBIAD1蛋白构象发生改变,不能与细胞中GGpp结合,无法受其调控从UBIAD1-HMG CoA还原酶复合体中解离,因此一直定位于内质网膜,同时HMG CoA还原酶无法从内质网膜上解离,导致其不能通过蛋白酶体降解,从而在内质网膜大量积累,其作为胆固醇和非甾醇类异戊二烯生物合成步骤中的关键限速酶不能被及时降解,造成细胞中胆固醇和非甾醇类异戊二烯的大量合成并积累。胆固醇和非甾醇类异戊二烯的生物合成见图2[44-48]。

图1 甾醇促进的内质网降解相关的HMG CoA还原酶途径中UBIAD1的功能。

图2 细胞中胆固醇和非甾醇类异戊二烯生物合成通路。
4小结
SCCD是一种稀有的常染色体显性遗传病,其临床表现为角膜随年龄增长逐渐混浊化,SCCD在男女中患病几率均等,因为胆固醇和结构脂类在角膜中的异常积累。该疾病于1924年首次报道[15],瑞士眼科医生Schnyder[16]阐明了本病的临床表现与遗传特性。直至2007年SCCD的分子基础才被确证[2-3]。SCCD的病因学基础为眼角膜中脂质的异常积累,致病机理的阐明为该疾病的分子诊断、靶向药物研发和特异性治疗提供了理论支持。Schumacher等科学家探究了UBIAD1与HMG CoA 还原酶的作用机制,详细描绘了UBIAD1-HMG CoA 还原酶在细胞中的角色与功能,为SCCD致病机理的阐明奠定了分子基础[44-48]。
UBIAD1基因突变可引起SCCD,但其在膀胱、前列腺等器官中可以作为抑癌因子,同时保护心血管系统免受氧化压力损伤。此外,UBIAD1也是人体中MK-4生物合成酶,在机体发育、脂质代谢和氧化损伤等方面中起到至关重要的作用,其它功能及作用机制仍需进一步阐明。
猜你喜欢
食品安全导刊(2022年32期)2022-12-07
上海化工(2022年3期)2022-06-30
昆明医科大学学报(2021年5期)2021-07-22
宁夏医学杂志(2020年3期)2021-01-21
中国化工贸易·上旬刊(2020年5期)2020-09-10
中国粮油学报(2017年5期)2017-07-19
橡塑技术与装备(2016年21期)2016-02-25
橡胶工业(2016年5期)2016-02-24
中国酿造(2014年9期)2014-03-11
中国粮油学报(2014年8期)2014-02-06
