
MOFs在催化CO2环加成反应中的催化位点类型
2020-06-04 09:51魏娜,王画,赵震,3
沈阳师范大学学报(自然科学版) 2020年2期
魏 娜, 王 画, 赵 震,3
(1. 沈阳师范大学 化学化工学院, 沈阳 110034;2. 沈阳师范大学 能源与环境催化研究所, 沈阳 110034;3. 中国石油大学 重质油国家重点实验室, 北京 102249)
0 引 言

图1 CO2环加成反应方程式Fig.1 Cycloaddition of epoxides and CO2 into cyclic carbonates
作为一种温室气体,大气中过量的CO2对地球的气候造成了严重的威胁,因此,CO2的固定与转化已经成为科学家们的研究热点。CO2与环氧化物环加成制备环状碳酸酯(图1)作为一种有效的转化途径,一直以来都备受科学家们的关注[1]。一方面,CO2作为合成化学中的一种化工原料,具有无毒、来源广且可再生的优点[2-4]。另一方面,产物环状碳酸酯是工程塑料的原料,也是精细化工制造业的中间体[5-6],展示出了非常高的工业价值。因此,该反应在环保及工业等领域都具有十分重要的研究意义。
CO2环加成反应的机理一般理解为,环氧化物上的氧原子首先与Lewis酸性位点作用,在亲核试剂的进攻下环氧化物开环,最后与CO2加成合环得到环状碳酸酯(图2)[7]。因此,Lewis酸性位点和亲核试剂是CO2环加成反应能够顺利进行的2个重要因素。目前,设计开发该反应的催化剂一般有2种思路,一是合成出具有Lewis酸性的材料,再加入亲核性的助催化剂来共同催化该反应,再者就是构建出同时展示出Lewis酸性和亲核性的催化材料。
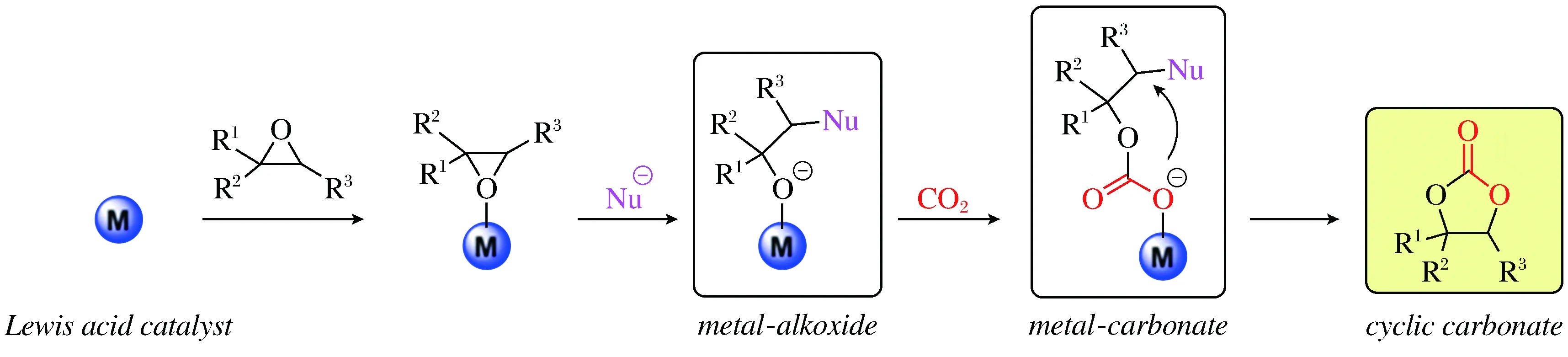
图2 环氧化物与CO2环加成制备环状碳酸酯反应机理Fig.2 Proposed mechanism for the cycloaddition of epoxides and CO2 into cyclic carbonates
金属有机骨架(metal-organic frameworks, MOFs)是由无机金属(簇)与有机桥连配体自组装而成的。基元化的结构使其展示出了结构和性能的多样性及可设计性[8-10],因此一直都是化学和材料学领域的研究热点[11-12]。MOFs材料在气体存储与分离[13]、传感[14]、质子导体[15]、生物医药[16]以及催化[17]等领域都具有良好的应用前景。作为一种催化材料,MOFs具有明显的优势。第一,催化位点来源广泛,金属节点、配体以及客体物种都可作为催化位点,并且可以于同一骨架中协同作用[18];第二,MOFs较大的比表面积和多孔结构均有利于反应进行,特殊的孔道(腔)结构也可以实现择形催化[19];第三,MOFs作为催化剂,易于与反应体系分离,回收简单,具有很好的可循环利用性[20]。
在CO2环加成反应中,MOFs结构中配位不饱和的金属节点是潜在的Lewis酸性位点,可以作为启动该反应的第一步。同时,通过对MOFs结构单元的选择或后合成修饰[21],可以引入功能性基团来增强其对CO2环加成反应的催化性能。因此,MOFs对于CO2环加成反应来说是一种非常有潜力的催化材料[22-24]。本文对近期MOFs材料催化CO2环加成反应的相关研究进行了综述,着重从金属催化位点的类型出发,结合催化机理来探讨MOFs作为催化剂在CO2环加成反应中的构效关系。
1 单金属位点
金属中心的Lewis酸性与其得电子能力有关。一般,原子半径大,金属性较弱的金属位点Lewis酸性较强[22]。然而,在实际进行催化时金属中心并非单独作用,配位原子的种类、孔道的结构等也会对催化能力产生一定影响。
2014年,Kathalikkattil等[25]设计合成了一种含有未配位羧基的MOF,{Cu(Hip)2(Bpy)}n(CHB)。相比于其他环氧化合物,CHB对烯丙基缩水甘油醚(AGE)与CO2环加成反应的催化活性更高,这是由于在该材料中,单原子Cu(Ⅱ)中心是Lewis酸性位点,而环氧化物环上的氧原子与Cu(Ⅱ)中心作用的同时,结构中未配位的羧基与AGE的侧链醚氧也产生了氢键作用,对该反应起到了促进的作用。因此,该工作启示人们可以通过巧妙的设计或选择MOF结构单元,向材料中引入适当的活性位点和官能团来提高其催化活性,这也为MOFs材料的合成提供了目标功能为导向的设计思路。
主族金属半径小、电荷高,对电子的吸引力强,因此,MOFs中的主族金属位点的Lewis酸性更强。同时,主族金属的原子轨道特性以及软硬酸碱理论均能证明,主族金属与有机配体合成的MOFs材料通常都会展示出很高的稳定性[26-27]。
2019年,一系列具有双次级结构单元(secondary building units, SBUs)的In-MOFs被合成出来,InDCPN-X(X=Cl、Br、I)[28]。In(Ⅲ)中心暴露出配位未饱和位点,且其末端配位的卤素离子(X=Cl、Br、I)可以作为弱碱性极化基团,对CO2分子产生极化和活化作用(图3)。因此,这些材料即使在相对温和的条件下(80 ℃,1 atm CO2)也可以对环氧化物的CO2环加成反应展示出高效的催化能力。通过对In(Ⅲ)中心上所修饰的卤素进行调变,材料中金属中心的酸性、极化基团的碱性、1D孔道的直径以及孔体积等结构性质均随之产生变化,这些都很好地证实了MOFs材料性能的调节可以通过对其SBUs的设计与改性来实现。该工作也为设计合成高效催化CO2环加成反应的Lewis酸/碱MOFs提供了一种新途径。

图3 InDCPN-X(X=Cl、Br、I)结构中的催化位点(a)及其催化CO2环加成反应机理(b)
稀土金属原子半径大,具有高配位数的特性[29],通常都处于配位未饱和状态,因此能够接受电子的外层空轨道比较多,具有较强的Lewis酸性,展现了很好的催化潜力。2017年,Xue等[30]设计合成了Gd-MOF材料。该材料在亲核试剂TBAB的协同催化作用下,对一系列环氧化物的CO2环加成反应均有很好的催化活性。这也说明,合成的Gd-MOF在各种反应中激活环氧化合物方面有潜在的应用前景。这也使更多的稀土金属型MOFs材料可以被考虑设计成具有高效催化效果的多相催化剂。
2 金属簇位点
在MOFs的形成过程中,由于配体构型的导向作用以及金属水解效应等因素,金属节点多以多核簇的形式存在[31],其作为催化位点的性能除了受自身性质影响之外,还受簇原子个数以及空间效应等影响。
2.1 双核簇
轮桨型金属簇M2(CO2)4是双核金属簇中最常见的类型。其中,2个金属中心通过4个配体羧基双齿桥联,每个金属中心又与4个羧基氧配位,通常在轴向方向与溶剂分子通过弱配位作用结合,通过活化处理可以除去,因此,M2(CO2)4簇中每个金属中心都是潜在的Lewis酸性位点。
经典的HKUST-1(Cu3(BTC)2)就是由轮桨型双核Cu簇为SBUs与均苯三甲酸构筑而成的。2012年,Eugenia等[32]对它催化环氧氯丙烷的CO2环加成反应性能进行了研究。不加助催化剂和溶剂,在100 ℃,0.7 MPa CO2下反应4 h,氯丙烯碳酸酯的产率仅为33%。这说明活化后的MOF暴露出了Lewis酸性Cu(Ⅱ)位点,对该反应有一定的催化能力,但是亲核试剂的缺失大大妨碍了反应的进行。
Zhou等[33]在2015年报道了一种基于非对称双核Ni簇的MOF材料Ni-TCPE1。在这个非对称的Ni2簇中,一个Ni(Ⅲ)中心分别与4个配体羧基、一个μ2-O和一个水分子配位,而另一个Ni(Ⅲ)中心则分别与2个配体羧基、一个μ2-O、2个水分子和一个DMF分子连接。经活化除去配位溶剂后,暴露出来的Lewis酸性Ni(Ⅲ)位点均匀分布在1D的纳米管壁上,且纳米管结构较大,使该材料对大尺寸的环氧化物与CO2环加成反应展示出了很好的催化能力,在100 ℃,1 MPa CO2的条件下催化氧化苯乙烯和苯基缩水甘油醚反应12 h的TON值高达2 000。
2.2 三核簇
三角形三核簇是MOFs中较为经典的一类金属簇节点。其中,3个金属中心通过一个μ3-O相连,形成的M3O簇又与6个配体羧基相连形成一个6连接的节点(M3(μ3-O)(CO2)6)。每个金属离子上通常都会有配位的溶剂分子,均为潜在的Lewis酸性位点。经典的MIL-88和MIL-101等均由三角形三核簇构筑而成。
2013年,MIL-101(Cr)对CO2环加成反应的催化能力被Zalomaeva等[34]进行了系统地研究。材料首先经过活化,除去三核Cr簇上的弱配位溶剂分子,暴露出不饱和配位点,从而展示出Lewis酸催化活性。催化实验结果表明,在25 ℃和0.8 MPa CO2条件下,MIL-101(Cr)在TBAB的共同作用下,环氧丙烷和氧化苯乙烯均可实现高效转化,对应环状碳酸酯产物的产率也可达82%和95%。但对于位阻较大的环氧环己烷的催化活性较低。
2017年,Wei等[35]利用二羧酸配体对苯二甲酸(H2BDC)和2,6-萘二甲酸(H2NDC)与稀土金属构筑了一系列12连接的、具有hcp拓扑构型的三维RE-MOFs(RE=Y, Er, Tb)。这些材料展示出了类MIL-88的结构,但具有更高的连接数。这些材料在较温和的条件下,对于CO2环加成反应均展示出了很好的催化活性,并且在循环催化5轮反应后,催化活性没有明显降低。这是因为结构中的七配位稀土金属中心处于配位未饱和状态,可作为Lewis酸性位点对该反应进行催化。此外,稀土元素的高配位数也使该材料具有高连接性,从而增强了材料的稳定性。
2.3 四核簇
四面体型的Zn4O簇是四核金属簇基MOFs中一种非常具有代表性的节点[36-37]。Zn4O簇中的4个Zn由一个μ4-O连接形成四核簇,每个Zn均为正四面体构型四配位(1个μ4-O和3个羧基氧),展示出了Lewis酸性。
2009年,Song等[38]研究了MOF-5催化CO2环加成反应的性能。实验结果表明,在50 ℃,0.2 MPa CO2,MOF-5中Lewis酸性的Zn4O簇与助催化剂TBAB协同作用下反应4 h,环氧丙烷的环加成产物碳酸丙烯酯的产率达到95%,并且在循环催化3次后催化活性和选择性没有明显下降。进一步地实验也证实了MOF-5在温和条件下对其他环氧化物也展示出了良好的催化能力。
平面四边形是四核金属簇中另一种比较常见的构型[39-40]。Gao等[41]在2019年合成了一种Ni-MOF材料。该材料具有三维多孔结构,结构中的孔道周围均匀分布着Ni4簇,Ni4簇中每个Ni(Ⅲ)中心上都有配位的水分子,经活化处理后暴露出Lewis酸性位点,因此该孔道结构可以作为CO2环加成催化反应的纳米反应器。催化实验也证明,在一定条件下,该材料对于一系列环氧化物的环加成反应均展示出了良好的催化效果。
2.4 六核簇
常见的六核金属簇多为八面体构型,其中6个金属原子通过8个μ3-O相连形成八面体M6O8簇。通过调节合成条件,该M6O8簇可以展示出6、8、10、12等多种配体连接数。
2013年,Kim等[42]对UiO-66和UiO-66-NH2催化CO2环加成反应的性能进行了研究。2种MOFs通过活化后都暴露出Lewis酸性Zr位点,但UiO-66-NH2由于骨架结构中存在NH2-基团,因此同时具有Lewis酸性和碱性,这也使UiO-66-NH2具有更好的催化活性。在2.0 MPa和100 ℃,氯苯做溶剂的条件下反应1 h,UiO-66-NH2催化SO的转化率达70%,而UiO-66催化SO的转化率仅为48%。对于其他环氧化物,UiO-66-NH2也具有高效的催化能力。
Lü等[43]在2019年设计合成了多种同构的8连接的M6O8簇基MOFs,M-NU-1008(M=Zr、Hf、Ce、Th)(图4)。尽管Lewis酸性测试表明Th>Zr>Hf>Ce,但Ce-NU-1008却展示出了最优的催化能力。这表明在M-NU-1008催化时,导致催化性能差异的并非这些元素自身的Lewis酸性差别。在这些催化剂中,M6簇作为潜在的Lewis酸性位点,在催化之前要经过活化除去配位水分子,而配位水分子除去的难易程度则决定了这些材料在催化过程中的Lewis酸性的强弱。经研究发现,Ce-NU-1008中M6簇的配位水分子最易除去,因此其展示出来的催化活性也最高。

图4 4种M6簇及M-NU-1008的结构Fig.4 The structure of M6 cluster and M-NU-1008
通过以上2个工作可以看出,在催过CO2环加成反应进行的过程中,影响催化效果的因素有很多,包括催化剂的结构特性、催化活性位点的性质、催化活性位点得到的难易程度等等,而决定性因素的确定,则有助于理解催化结果与机理,对高效催化剂的设计合成有很大的导向性作用。
2.5 其他核数金属簇
2017年,Wei等[44]首次报道了2种五核Yb(Ⅲ)簇的MOFs材料,Yb-DDPY和Yb-DDIA。在这2种材料的结构中,Yb(Ⅲ)中心均处于配位不饱和状态。这些Yb5-MOFs在60 ℃和1.0 MPa CO2条件下,对CO2环加成反应均展示出了良好的催化能力。同时,Yb-DDIA由于配体上存在着未配位的-COOH基团,能作为Brønsted酸位点协同Lewis酸Yb(Ⅲ)中心进行催化,因此展示出了更好的催化活性。
Eddaoudi等[45]在2014年报道了一种由九核Y(Ⅲ)簇([Y9(μ3-OH)8(μ2-OH)3(O2C-)18])和三角形三羧酸配体组装形成的(3,18)连接网络MOF,gea-MOF-1。在Y9簇中,6个8配位的Y(Ⅲ)中心和3个6配位的Y(Ⅲ)中心,均为Lewis酸性位点。加之该材料的大空腔结构使得这些开放金属中心催化位点更易与反应物接触作用。因此,gea-MOF-1与TBAB协同作用,在120 ℃和2 MPa CO2的条件下,能高效催化CO2环加成反应。
3 异双金属位点
2018年,Dae-Won Park课题组[46]设计合成了Ni-Co-MOF。骨架节点SBUs同时含有Ni(Ⅱ)和Co(Ⅱ)这2种Lewis酸性位点,Ni(Ⅱ)与Co(Ⅱ)中心的耦合作用使二者间发生电荷转移,协同催化CO2的转化,比单金属位点Ni-MOF和Co-MOF具有更好的催化活性。在80 ℃,1.2 MPa CO2的条件下,Ni-Co-MOF的催化转化率及产物选择性均达到了90%以上。
同年,Tang等[47]设计合成了双金属Au/ZnMOF中空纳米笼(RH Au/ZnMOF和RD Au/ZnMOF)(RH正六面体;RD菱形十二面体)。该中空笼结构的壳壁上分布着高分散性的强酸性位点,在温和的反应条件下可高效催化CO2环加成反应。结构中Au(Ⅲ)和Zn(Ⅱ)这2种位点的协同作用以及该材料独特的中空结构,使其比ZIF-8以及均相的金属催化剂具有更高的催化活性。
由此可知,向MOFs中引入不同金属原子使其产生耦合作用,同时开发更多的配位不饱和金属位点,是设计高性能异相MOFs催化剂的一种行之有效的方法。
4 其他金属位点
由于MOFs结构的可设计性,可以通过向其配体或者空腔中引入同种或其他金属离子来实现双金属位点MOFs催化材料的合成。
2014年,Gao等[48]设计合成了一种含氮杂大环的四羧酸配体,并利用它合成了一种与MOF-505具有相似拓扑结构的MOF,MMCF-2。在该MOF中存在2种金属位点,一种是作为骨架节点的轮桨型双核Cu簇,另一种就是在合成过程中嵌入配体上氮杂大环的单原子Cu位点。在催化剂量相同时,MOF-505上只含有双核Cu簇金属位点,因此,MMCF-2的Lewis酸位点更多,催化能力更强。同年,该课题组又报道了一种由八羧酸卟啉配体构筑的MOF,MMPF-9[49]。该MOF高密度的Lewis酸活性位点使其在室温常压下也能够高效催化CO2环加成制备环状碳酸酯。可见,通过对MOF配体的设计,向骨架中引入高密度的催化活性位点,是开发高效异相催化剂一种有效且适用范围广泛的途径。
2017年,Liu等[50]通过“ship in a bottle”的方法,原位将salen化合物(salen-Cu(Ⅱ))封装到MIL-101(Cr)的笼型空腔内,得到了一种双活性位点(Cr(Ⅲ)和Cu(Ⅱ))的MOF催化剂salen-Cu(Ⅱ)@MIL-101(Cr)。CO2环加成催化实验表明,在相同反应条件下,含双Lewis酸中心的salen-Cu(Ⅱ)@MIL-101(Cr)的催化活性明显高于单独的salen-Cu(Ⅱ)和MIL-101(Cr)。在室温常压下salen-Cu(Ⅱ)@MIL-101(Cr)催化碳酸丙烯酯的产率可达87.8%。但在催化过程中MIL-101空腔中的salen-Cu(Ⅱ)有部分流失,导致循环使用时催化剂的催化活性略有下降。
2019年,一种骨架封装Ni纳米粒子的MOF复合材料被合成出来,其催化CO2环加成反应的能力被系统地探究了[51]。Singh等通过后合成手段设计得到了一种含有Ni0和Zr4+这2种金属位点的Ni@ZrOF材料。因为Ni纳米粒子的引入,使得该复合材料对CO2的吸附能力得到了很大的提高。同时,在催化反应过程中,Lewis酸性Ni0和Zr4+位点均可起到活化环氧化物上O原子的作用。因此,对于CO2环加成反应,Ni@ZrOF比未封装Ni纳米粒子的UiO-66-NH2具有更高的催化活性。
综上工作表明,将具有催化活性的物种封装入骨架中可以大大提高材料的催化能力,这为未来制备具有高效催化性能的MOFs材料提供了一种可能的方向。
5 结论与展望
MOFs材料具有潜在的Lewis酸性金属中心及CO2吸附能力,因此成为了一种催化CO2环加成反应非常有潜力的材料,但在实际应用中仍然面临一些问题。大部分MOFs因为只有金属节点作为Lewis酸性位点,所以通常需要加入亲核性助催化剂来进行协同催化,因此造成了资源浪费、产物分离困难等问题。另一方面,MOFs是通过结构单元间的配位作用组装而成的,相对于分子筛、金属氧化物等传统催化材料,它的结构稳定性较差。因此,设计开发出具有单组分、高效催化性能且廉价、稳定的MOFs材料,是MOFs催化性能研究人员们的共同目标。
感谢沈阳师范大学优秀人才支持计划资助项目(51300425)。
猜你喜欢
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
云南化工(2020年11期)2021-01-14
装备维修技术(2020年5期)2020-11-20
矿产综合利用(2020年1期)2020-07-24
无机化学学报(2020年7期)2020-07-20
当代陕西(2019年6期)2019-04-17
分析化学(2017年9期)2017-10-16
科技创新导报(2016年30期)2017-03-15
外语学刊(2014年3期)2014-12-03
郑州大学学报(理学版)(2013年2期)2013-03-11
