
自噬调控多功能蛋白p62/SQSTM1参与肿瘤及其微环境的研究进展
2020-05-23 08:12:00陈佳锋傅修涛丁振斌
中国临床医学 2020年2期
陈佳锋, 傅修涛, 丁振斌
复旦大学附属中山医院肝脏外科,上海 200032
细胞中蛋白质的降解过程主要由泛素-蛋白酶体系统和自噬-溶酶体系统完成。自噬(autophagy)能够调控各种生理和病理过程,甚至影响整个机体的代谢。自噬与肿瘤的关系错综复杂,其中自噬接头蛋白p62/SQSTM1(以下简称“p62”)在肿瘤及其微环境中起到了重要作用。本文就自噬及多功能蛋白p62参与肿瘤及其微环境调控机制的研究进展作如下综述。
1 自噬
自噬是真核细胞中进化上高度保守的,借助溶酶体将胞浆内代谢产物及受损细胞器降解成氨基酸、核苷等小分子并重新循环利用的生物学过程。根据自噬进入溶酶体的方式,可将自噬分为3种类型:巨自噬、微自噬和分子伴侣介导的自噬(chaperon-mediated autophagy,CMA)[1]。通常意义的自噬即巨自噬,最主要的特征是形成杯状双层膜结构包裹未折叠蛋白质、受损细胞器,然后经过核化、延长,形成自噬小体,最终与溶酶体融合形成自噬溶酶体以降解内膜及其包裹的底物[2]。
自噬根据底物的特异性和转运机制的不同,可分为非选择性自噬和选择性自噬。前者非选择性降解胞内大分子物质及细胞器并循环利用氨基酸等原料,这个过程受到细胞能量感受器AMPK和mTORC1的调控[3]。选择性自噬,如线粒体自噬、过氧化物酶体自噬等,能在特定条件下清除受损细胞器,缓解内质网及线粒体氧化应激压力,维持细胞的完整性和功能,抑制基因突变从而抑制肿瘤的发生。选择性自噬需要自噬接头蛋白或者其他靶向溶酶体的蛋白桥接特异性底物和自噬受体LC3(ATG8)。p62、NBR1、NDP52、Nix、Cb1、Stbd1、OPTN等均是含有人微管相关蛋白轻链识别序列(LC3-interacting region, LIR)结构域的自噬接头蛋白,其中p62是最早被研究发现的自噬接头蛋白[4]。
2 p62的分子结构及相关信号通路
2.1 p62的分子结构及细胞定位 自噬接头蛋白p62已明确的作用域包括PB1结构域(Phox and Bem1p)、ZZ型锌指结构域(Zinc finger)、TB结构域(TRAF6-binding domain)、LIR结构域、KIR结构域(Keap1-interacting region)和UBA结构域(ubiquitin-associated domain)[5],见图1。p62蛋白N端的PB1结构域与p62的低聚反应有关,该结构域同时与PKCζ、MEKK3、MEK5、ERK1和NBR1等序列相互作用,进而调控PKCζ-JNK-caspase3凋亡通路等;TB结构域和ZZ型锌指结构域可以结合泛素连接酶(TNF receptor associated factor 6,TRAF6)调控肿瘤坏死因子TNF-α及其下游信号通路;LIR结构域则结合自噬泡上微管相关蛋白1轻链3(LC3),是自噬过程的重要环节;KIR结构域则与Keap1-Nrf2信号通路有关;而C端的UBA结构域则参与蛋白酶体降解以及选择性自噬的生理过程。正因为p62有多个功能的结构域,其在细胞凋亡、炎症反应、细胞生存、信号转导以及肿瘤进展等方面均起到了重要的调控作用[6]。
研究[7-8]发现,p62不仅广泛存在于细胞质和细胞核,也分布于自噬体和溶酶体上。在应激环境下,该蛋白也可以定位于蛋白质聚集体、受损线粒体以及细胞内入侵的病原体上。
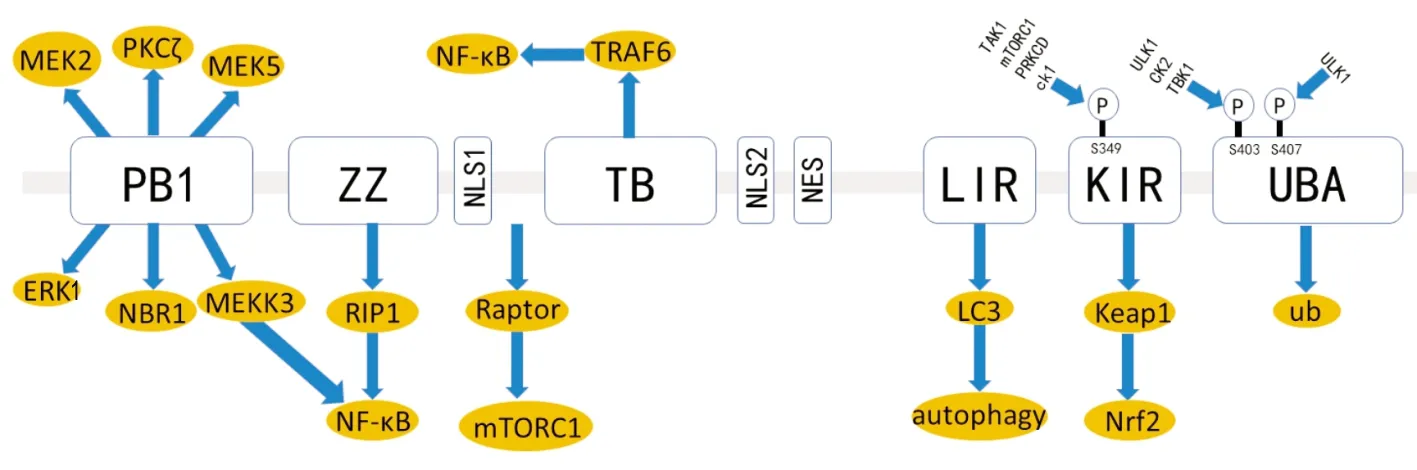
图1 p62的分子结构及作用域
PB1:Phox and Bem1p结构域;ZZ:ZZ型锌指结构域;NLS1:核输出信号1;TB:TNF受体相关因子6绑定域;NLS2:核输出信号2;NES:核定位信号;LIR:人微管相关蛋白1轻链3识别序列;KIR:Keap1作用域;UBA:泛素化相关域
2.2 p62相关信号通路 p62作为信号枢纽,参与激活mTORC1、NF-κB、PKCζ-JNK-caspase3和Keap1-Nrf2等多条重要信号通路。
(1)Keap1-Nrf2信号通路:研究[9-10]发现,ser349位点磷酸化的p62通过KIR结构域竞争性结合Keap1,释放Nrf2进入细胞核内激活下游AREs和EpREs等反应元件表达,从而发挥抗氧化效应。而多种恶性肿瘤中广泛存在Nrf2的持续激活,可通过调节谷氨酰胺分解、嘌呤核苷酸和谷胱甘肽合成以及磷酸戊糖途径,促进肿瘤生长[11-12]。
(2)NF-κB信号通路:p62通过PB1结构域结合RIP,激活TNF-α介导的NF-κB信号通路,或者通过TB结构域结合TRAF6直接激活NF-κB通路[13]。该通路促进活性氧簇(reactive oxygen species,ROS)的产生,进而促进DNA损伤和基因突变,同时也可促进肿瘤细胞增殖和抑制p53介导的细胞凋亡。
(3)mTORC1信号通路:p62能够与Rag、Raptor以及TRAF6蛋白相互作用增加mTORC1的活性,继而激活S6K1、4EBP、ATF4等激酶,增强合成代谢反应,促进肿瘤细胞增殖[14-15]。同时,mTORC1持续激活上调癌基因c-Myc的活性,激活肿瘤相关纤维母细胞,促进转化生长因子β(TGFβ)等的合成,从而促进肿瘤的形成[16]。
3 p62与自噬
研究[17-18]表明,p62蛋白可通过UBA和LIR结构域分别结合泛素化底物及自噬小体,将待降解底物转运至自噬溶酶体系统。生理条件下,p62蛋白分子的UBA结构域形成稳定的二聚体,与泛素的亲和力较低。但在泛素过表达、热休克、蛋白酶体抑制剂等诱导下,该分子UBA结构域的Ser407位点被ULK1激酶磷酸化,激活成为单体。随后,酪蛋白激酶-2或者TANK结合激酶-1磷酸化UBA结构域的Ser403位点,p62与泛素的结合力显著增强。p62蛋白泛素化干扰UBA结构域的二聚化,从而恢复UBA结构域识别泛素化底物的效能,促进选择性自噬的启动[19]。在选择性自噬中,p62不仅作为泛素化蛋白的接头蛋白,也作为底物被自噬过程降解。因此,在生理条件下基础水平的自噬可以维持p62处于一个较低的浓度水平。此外,NBR1和ALFY这2种与p62相互作用的蛋白同样参与自噬过程,p62蛋白分子PB1结构域能够结合NBR1,加速自噬对泛素化底物、蛋白聚集体、受损细胞器的降解,ALFY促进p62阳性聚合物的装配进行自噬降解[20-21]。
4 p62与肿瘤及其微环境
4.1 p62与肿瘤 多功能蛋白p62涉及多种疾病的发生发展,包括佩吉特骨病、肌萎缩侧索硬化及额颞叶变性等均与SQSTM1基因突变有关。研究[22-25]证明,肝细胞癌、胰腺癌、肾癌以及结直肠癌等多种肿瘤中均发现了p62聚集,并易在胞质内形成富含p62蛋白和泛素的马洛里小体(Mallaory body,MB)和hyaline globule等包涵体,提示高水平的p62与肿瘤形成密切相关。Umemura等[15]研究发现,肝细胞癌的发生发展可能与p62激活Nrf2、mTORC1和c-Myc介导的通路有关,而且p62的聚集和肝癌术后的早期复发相关。Duran等[26]研究提示,p62敲除的小鼠中,RAS诱导的细胞转化、肿瘤生成及NF-κB活性均受到抑制,同时细胞内ROS增强、凋亡增加,因此肺腺癌的发生率明显下降。不仅如此,p62也是胰腺癌进展的重要驱动因子之一,在RAS诱导的胰腺导管腺癌小鼠中,NF-κB通路激活诱导p62的合成并作用于TRAF6,正反馈增强NF-κB信号通路,促进肿瘤进展。p62的聚集同时激活Nrf2信号通路,诱导癌基因MDM2表达,促进胰腺导管癌细胞的生长[22]。肿瘤干细胞是肿瘤治疗后复发的重要原因之一。研究[27]发现,在乳腺癌肿瘤干细胞中,p62表达显著增高,并且p62可以促进MYC的转录,从而影响乳腺癌肿瘤干细胞以诱导肿瘤复发和耐药。
p62不仅在胞浆内发挥作用,也可调节细胞核内的转录过程,故p62也被视作转录调控蛋白之一,但细胞核内p62的功能尚不明确。研究[28]发现,细胞核内的p62水平与肺鳞状细胞癌肿瘤分化程度相关,但与其他病理学或临床指标没有明显相关性。在黑色素瘤中,FERMT2 mRNA是肿瘤转移及预后不良指标,核内p62的PB1结构域可以和RNA结合蛋白IGF2BP1相互作用,继而延长FERMT2半衰期,促进黑色素瘤的进展[29]。最新的一项食管癌研究[30]显示,核内p62表达水平与化疗的敏感性相关。新辅助化疗和手术后,胞质内p62蛋白增加以及核内p62蛋白水平降低均提示预后不良。
4.2 p62和肿瘤微环境 肿瘤曾被认为是一种由于基因表达失调导致的细胞疾病,然而众多研究[31-35]证实,肿瘤微环境(tumor microenviro-nment, TME)在肿瘤的发生、发展、转移以及对肿瘤治疗的抵抗方面都起到了重要作用。Valenci等[32]研究发现,与肿瘤细胞中的表达相反,p62在肿瘤基质(尤其是肿瘤相关成纤维细胞)中表达水平下调。深入研究[32-33]结果显示,p62表达缺陷的纤维母细胞分泌更多IL-6,促进TGFβ的合成,诱导肿瘤相关成纤维细胞向肌成纤维细胞转化,进而促进肿瘤的发展。同时成纤维细胞p62的缺陷导致c-Myc水平降低,下调还原型谷胱甘肽合成,氧化应激增强,进一步促进IL-6和TGFβ表达。前列腺癌中,p62缺陷的纤维母细胞抑制mTORC1/c-Myc信号通路激活,促使代谢重编程,IL-6和TGFβ分泌增加,细胞间质ROS增多,炎症反应被激活,促进前列腺肿瘤细胞的增殖和侵袭。Linares等[34]研究表明,基质细胞p62表达缺陷,能够诱导ATF4的聚集,启动下游转录途径,转运天冬酰胺到肿瘤细胞中,维持肿瘤在营养缺乏的微环境下生存,同时激活mTORC1,进一步促进肿瘤发展。正常肝脏细胞中,p62蛋白能够诱导VDR信号通路,抑制肝星状细胞(hepatic stellate cell,HSC)的功能,抑制p62后,活化的HSC诱导肝脏炎症反应及纤维化,支持肿瘤进展[35]。总之,p62能够诱导肿瘤微环境代谢重编程,调节细胞因子分泌,从而发挥微环境调控肿瘤生长的功能。而抑制mTORC1的抗肿瘤治疗的效果,会因基质中mTORC1失活产生的效应而降低,因此针对p62的靶向治疗需要平衡其各方面的副作用。
4.3 自噬、p62与肿瘤及其微环境 p62介导的选择性自噬及NF-κB等肿瘤刺激信号通路的激活可能是肿瘤形成的始动因素之一。在人EBV阳性B淋巴瘤细胞中,EBV感染产生的活性氧/活性氮(ROS/RNS)激活Keap1-Nrf2信号通路,诱导p62的过度累积以及介导的选择性自噬,促进蛋白酶体对DNA修复蛋白CHK1和RAD51的降解,诱导肿瘤的形成[36]。多项研究[15,37-38]显示,自噬缺陷不仅促进细胞内受损DNA的累积,染色体断裂和不稳定,而且其引起的p62聚集能使NF-κB、mTORC1和Nrf2信号通路表达上调,诱发肝细胞癌形成。DEAD box蛋白5(DDX5)上调自噬水平后,可促进p62的降解,抑制肿瘤形成。而miR-17-5p可以上调DDX5,DDX5与p62结合后,干扰p62/TRAF6介导的mTOR信号通路的激活,进而延缓甚至阻止慢性肝病向肿瘤的进展[39]。细胞周期蛋白D1(cyclinD1)能够促进细胞DNA合成及分裂。Wu等研究[40]证实,通过p62介导的对原癌基因miR-224和cyclinD1蛋白的选择性自噬降解作用,能够抑制肝细胞癌形成及进展。另外,研究[23]表明,SQSTM1基因高表达与肾透明细胞癌的发生相关,其表达产物p62蛋白促进了NF-κB、mTORC1和Nrf2信号通路。
在自噬起始复合物ULK1-ATG13-FIP20-ATG101形成受阻后,p62仍可以促进下游NF-κB通路维持肿瘤发展,因此p62和自噬协同维持肿瘤生长。p62的S351位点磷酸化可增强其在选择性自噬过程中结合Keap1的能力,从而诱导Nrf2信号通路持续表达,改变肿瘤细胞代谢满足其生长需要以及应对肿瘤微环境压力,促进肝细胞肝癌的发展[9]。Wei等[41]通过一系列研究证实,在乳腺肿瘤中敲除FIP200抑制自噬后,再敲除p62可以进一步抑制肿瘤的生长。但也有研究[42]报道,Flightless-1(Flil)蛋白与p62相互作用,阻碍p62介导的选择性自噬从而促进乳腺肿瘤进展。X连锁凋亡抑制蛋白(XIAP)是多种肿瘤的促癌蛋白,在乳腺和结肠肿瘤中,XIAP能够抑制p62的表达以及介导的选择性自噬,进而导致肿瘤的发展和转移[43]。研究[44]发现,在急性髓系白血病小鼠模型中,抑制p62可减少白血病细胞的增殖,阻碍白血病进展,而这归功于p62的缺陷导致的线粒体自噬功能失调以及肿瘤细胞能量失衡。
肿瘤转移涉及多步过程,包括肿瘤细胞获得侵袭能力,脱离原发部位,侵入血管成为循环肿瘤细胞,最后移出血管定植在转移部位。上皮来源的肿瘤细胞主要通过上皮向间质转化(EMT)获得侵袭、转移能力。Jiang等[45]研究发现,前列腺癌中p62提升HDAC6水平,降低α-微管蛋白的乙酰化以及微管的稳定性,最终在选择性自噬功能受损的情况下促进肿瘤EMT,增强癌细胞的增殖侵袭能力。与之类似,自噬抑制剂作用于RAS突变的肿瘤细胞后,p62集聚诱导NF-κB信号通路,继而促进肿瘤EMT[46]。最近的一项研究[47]发现,通过药物或者基因敲除等方法抑制自噬水平,能够诱导糖酵解调节因子Pfkfb3的异常表达,促进乳腺肿瘤干细胞从休眠状态转变至增殖状态,导致肿瘤复发。而激活自噬则可以使肿瘤干细胞处于休眠状态,这其中的机制与p62的UBA结构域与Pfkfb3相互结合,促进后者的降解有关。另一项乳腺癌研究[48]证实,肿瘤中过表达的TRIM59抑制p62介导的对PDCD10的选择性自噬降解,进而促进肿瘤的转移能力。SNAI1是EMT的重要驱动因子之一,在HeLa和H1299细胞中,SNAI1和LC3及p62相互作用,促进前者被自噬过程降解,从而阻碍SNAI1进入细胞核内,抑制与肿瘤侵袭相关基因的转录[49]。
p62及自噬协同调控肿瘤微环境及免疫状态。肿瘤基质中,p62可以介导RBPJ/CSL的自噬性降解,进而增加肿瘤相关成纤维细胞基因表达,维持周围肿瘤细胞的生长[50]。Zhong等[51]研究证实,p62缺陷的肿瘤相关巨噬细胞(TAM)线粒体自噬水平下降,导致受损线粒体以及NLRP3炎性小体的增加,刺激IL-1β和IL-18等细胞因子分泌,可以进一步促进肿瘤进展。在缺氧肿瘤微环境中,抑制自噬可以导致p62蛋白的累积并结合到活化的信号转导与转录激活因子3(pSTAT3),然后诱导泛素-蛋白酶体系统降解激活,从而提高免疫T细胞的肿瘤杀伤作用[52]。
p62及自噬的表达水平与肿瘤的化学治疗反应密切相关。多项研究[53-54]显示,耐铂类药物的卵巢上皮癌细胞内p62水平明显增高,这与p62激活NF-κB通路有关,而通过自噬诱导剂降低p62水平可以明显增加卵巢上皮癌对铂类的敏感性。Battista等[55]研究发现,5-氟尿嘧啶、顺铂以及多西紫杉醇耐药的人喉表皮癌细胞(TDR Hep-2)中p62-Nrf2通路水平明显增高,以对抗化疗药物产生的氧化应激反应以及蛋白毒性。在KIR、LIR、UBA结构域变异的p62的Hep-2细胞中,肿瘤对化疗药物敏感性增加。因此,Leidal等[56]认为,在治疗某些肿瘤的过程中,不仅要抑制自噬水平,需同时抑制p62的水平。
综上所述,细胞自噬及p62对于肿瘤是一把双刃剑,基础水平的自噬可以维持染色体的稳定性,抑制肿瘤形成,但在肿瘤形成后可以维持肿瘤进展的营养代谢需求。抑制自噬后,高水平的p62可以通过Nrf2、NF-κB和mTORC1等信号通路促进血管生成以及肿瘤细胞生长,而在基质细胞中低水平的p62则通过促进IL-6和TGFβ等分泌,使肿瘤细胞代谢重新编程以应对营养缺乏的微环境。由于p62复杂的功能结构域参与多个重要信号通路网络,同时影响自噬、细胞凋亡、免疫逃逸等多个生理病理过程,靶向p62药物的效果在治疗肿瘤过程中易受到肿瘤类型、分期、p62蛋白表达量以及其他药物的影响。故调节自噬的药物已在部分肿瘤中得到一定应用,但其对于治疗肿瘤的有效性仍有待进一步考证。因此自噬与p62相互调节以及在肿瘤调控方面的作用需要更多深入的研究,最终为靶向自噬及p62的抗肿瘤药物治疗提供理论基础。总之,当利用自噬系统进行肿瘤相关治疗时,检测肿瘤组织中p62的表达是一个较好的预后及治疗辅助指标,同时需要考虑p62与相关通路的相互影响,以实现最佳的肿瘤联合治疗。
猜你喜欢
第一财经(2019年8期)2019-08-26 17:53:46
广州大学学报(自然科学版)(2019年1期)2019-05-07 01:33:26
天津科技大学学报(2016年1期)2016-02-28 16:59:45
湖北师范大学学报(自然科学版)(2015年2期)2016-01-10 08:41:53
浙江大学学报(农业与生命科学版)(2015年4期)2015-12-15 12:47:42
哈尔滨医药(2015年2期)2015-12-01 03:57:13
学习月刊(2015年14期)2015-07-09 03:37:48
中国医学科学院学报(2015年5期)2015-03-01 04:03:46
物理化学学报(2015年5期)2015-02-28 17:34:47
现代检验医学杂志(2015年2期)2015-02-06 02:01:01
