
柱后还原-高效液相色谱法同时测定调制乳粉中维生素K1和维生素K2
2020-05-22 12:41孙珊珊公丕学张艳侠王明栋卢兰香李新玲刘艳明
色谱 2020年7期
孙珊珊, 公丕学, 张艳侠, 王明栋, 卢兰香, 李新玲, 薛 霞, 刘艳明*, 张 峰
(1. 山东省食品药品检验研究院, 山东 济南 250101; 2. 中国检验检疫科学研究院, 北京 100176)
维生素K又叫凝血维生素,是一类具有叶绿醌生物活性的萘醌类化合物,是肝内合成凝血酶原的必需物质,参与止血、骨代谢和细胞生长,当体内缺乏时可造成凝血障碍,还具有降血脂、软化血管、预防骨质疏松等诸多功能,是人体必需的脂溶性维生素[1,2]。天然存在的维生素K有维生素K1(植物甲萘醌,VK1)和维生素K2(甲萘醌类,VK2)两种,广泛存在于自然界中,与人体代谢密切相关,也是研究及应用最多的两类化合物[3,4]。维生素K1和维生素K2都是脂溶性维生素,对光和碱敏感,有一定的耐热性,在生物体内具有不同强度的生物活性[5]。维生素K1为单一化合物,其侧链上只包含有一个异戊烯结构单元,主要来源于绿叶蔬菜(如菠菜、甘蓝、莴苣等)与大豆及其制品[6]。维生素K2由肠道细菌合成,另外纳豆、蛋黄、动物肝脏、发酵品中少量含有[7],维生素K2是由一个甲基萘醌母核和一条C3位带有异戊二烯侧链组成的系列化合物,根据侧链上异戊二烯长短不同共有14种同系物,其中维生素K2(35)亦称为甲萘醌-7(MK-7),是维生素K2中功能最广泛、活性最强大、作用最持久、成分最安全的一种[8]。
维生素K1和维生素K2作为一种营养补充剂应用到调制乳粉中,对于维生素K缺乏症有很好的预防作用。而维生素K1是目前国内主要的营养强化剂,维生素K2现在处于刚发展阶段。2016年国家卫计委第8号公告中批准将维生素K2(35)作为食品营养强化剂在调制乳粉中添加使用[9],在美国、欧洲及我国台湾地区早已批准维生素K2作为营养补充剂添加,在各类食品中根据实际需要添加。因此目前国内维生素K2在食品中的应用相对较少。
国内对食品中维生素K1的检测研究较多,而食品中维生素K2的检测,维生素K1和维生素K2的同时检测方法相对较少。维生素K1和维生素K2的检测方法主要有分光光度法[10]、液相色谱-紫外检测法[11-13]、液相色谱-荧光检测法[14-17]、液相色谱-质谱法[18-20]等。其中分光光度法分析步骤繁琐,且精密度不高;高效液相色谱-紫外检测法选择性和灵敏度不高,无法满足食品中微痕量组分的检测需求;高效液相色谱-质谱法虽然具有较高的灵敏度,但其仪器复杂、成本较高,而且食品基质复杂,易影响定量准确性。高效液相色谱-荧光检测方法(HPLC-FLR)灵敏度高,特异性强,维生素K的不饱和酮结构共轭体系小,其本身并不能产生荧光,通过柱后锌粉还原使维生素K同系物的母核萘醌环发生荧光反应,产生荧光物质[21],从而提高其在高效液相色谱荧光检测上的灵敏度和选择性。
目前,液相色谱-荧光检测同时检测调制乳粉中维生素K1和维生素K2含量的方法鲜有报道。本实验采用柱后锌粉还原-高效液相色谱-荧光检测方法对调制乳粉中的维生素K进行研究,建立了定性定量准确可靠、灵敏度高、特异性强的一种高效液相色谱-荧光检测同时测定维生素K1和维生素K2的检测方法,对调制乳粉中营养强化剂维生素K1和维生素K2的检测、应用和监管提供技术支撑。
1 实验部分
1.1 仪器、试剂与材料
Waters高效液相色谱仪(带荧光检测器)、Xbridge C18色谱柱(150 mm×4.6 mm, 3.5 μm)(美国Waters公司);锌粉还原柱(50 mm×4.6 mm,上海安谱实验科技股份有限公司); 3-18K型冷冻离心机(德国Sigma公司); AB204-S型电子天平(德国Mettler Toledo公司); MS3型旋涡混合器(德国IKA公司); SB-800DTD超声清洗仪、SW22恒温水浴振荡器(中国宁波新芝生物科技股份有限公司); HGC-24型氮吹仪(中国恒奥科技有限公司); Milli-Q超纯水系统(美国Millipore公司)。
维生素K1标准品(德国Dr. Ehrenstorfer公司);维生素K2标准品(美国ChromaDex公司)。甲醇、正己烷、二氯甲烷(色谱纯,德国Merck公司);冰乙酸(色谱纯,天津市康科德科技有限公司);无水乙醇、氯化锌、氯化钠、乙酸钠、氢氧化钠(分析纯,国药集团化学试剂有限公司);碳酸钾(优级纯,天津市科密欧化学试剂有限公司)。脂肪酶(酶活力≥700 U/mg,美国Sigma公司);实验用调制乳粉购自超市。
1.2 标准溶液配制
分别准确称取维生素K1和维生素K2标准品各 0.010 0 g,置于10 mL容量瓶中,用正己烷溶解并定容,配制 1 000 μg/mL 维生素K1、维生素K2标准储备液,充分摇匀,在棕色试剂瓶中于-18 ℃冰箱中储存。
分别吸取一定量的维生素K1和维生素K2标准储备液,氮气吹干后,用甲醇稀释,混匀,配制成质量浓度为100 μg/mL 的混合标准溶液。将上述混合标准溶液用甲醇逐级稀释成 0.002 5、0.01、0.05、0.1、0.5、1.0和2.0 μg/mL 的混合标准工作液,现用现配。
1.3 样品前处理
称取2.5 g样品,置于50 mL离心管中,加入0.8 g脂肪酶,用10 mL温水溶解,涡旋混匀,于(37±2) ℃恒温水浴振荡,酶解4 h后,取出加入1 mL 2.5 mol/L 氢氧化钠溶液,涡旋30 s后,再加入10 mL无水乙醇,充分混匀,进行皂化反应。加入10 mL正己烷,涡旋提取5 min,以 6 000 r/min 离心3 min,移取上层正己烷层于另一个50 mL离心管中,再加入10 mL正己烷萃取一次,合并提取液,用10 mL饱和氯化钠溶液水洗有机相后氮气吹干,用甲醇2 mL复溶,过0.22 μm有机滤膜,待液相色谱测定。整个实验过程均避光操作。
1.4 分析条件
色谱柱:Xbridge C18色谱柱(150 mm×4.6 mm, 3.5 μm);柱后锌粉还原柱(50 mm×4.6 mm);柱温:35 ℃;流动相:甲醇溶液(含0.03%(v/v)冰乙酸、1.5 g/L 氯化锌和0.5 g/L 无水乙酸钠);流速1.0 mL/min;进样体积:10 μL;荧光检测:激发波长(λex)326 nm,发射波长(λem)432 nm。
2 结果与讨论
2.1 色谱条件的优化
2.1.1 荧光条件的优化
维生素K本身不带荧光,经锌粉还原后产生荧光物质进行检测。当有锌离子存在时,维生素K可以通过锌柱被还原后产生荧光物质,为了提高被测样品在高效液相色谱-荧光检测器上的响应值,提高灵敏度,必须选择合适的激发波长和发射波长。本文采用Waters 2475荧光检测器的三维(3D)扫描功能,对锌粉还原后的维生素K1和维生素K2混合标准工作液进行激发光谱和发射光谱扫描。
结果如图1a所示,固定发射波长为430 nm,在200~420 nm激发光谱下扫描还原后的维生素K1和维生素K2,得到二者的激发波长为243 nm和326 nm,其中还原后维生素K1和维生素K2在λex为326 nm时的响应值约是λex为243 nm时的1.5倍,因此选择λex为326 nm。如图1b所示,固定λex为326 nm,在336~610 nm波长范围内扫描发射光谱,得到最佳λem为432 nm。因此通过分析,选择最佳λex为326 nm,最佳λem为432 nm。

图 1 还原后维生素K1和维生素K2的(a)激发光谱和(b)荧光光谱图Fig. 1 (a) Excitation and (b) fluorescence spectra of the vitamin K1(VK1) and vitamin K2(VK2) after reduction a. emission wavelength (λem)=430 nm; b. excitation wavelength (λex)=326 nm.
2.1.2 色谱柱和流动相

图 2 维生素K1和维生素K2混合标准溶液(1.0 μg/mL)的色谱图Fig. 2 Chromatogram of the VK1 and VK2 in the mixed standard solution (1.0 μg/mL)
维生素K1和维生素K2脂溶性较强,具有弱极性,其在反相色谱柱上保留很强,容易导致峰宽拓展,峰形钝化,影响测定的灵敏度。本文采用含0.03%(v/v)冰乙酸、1.5 g/L 氯化锌和0.5 g/L 无水乙酸钠的甲醇溶液为流动相(pH为6.0±0.1),通过Xbridge C18色谱柱(150 mm×4.6 mm, 3.5 μm)进行分离,锌粉还原柱还原后荧光检测器检验。由图2可见,维生素K1和维生素K2的峰高、峰形较理想,出峰时间早,缩短了分析检测时间。
2.2 前处理条件的优化
本文以调制乳粉为研究对象进行前处理条件的优化考察,将一定量的含维生素K1和维生素K2的营养添加剂添加到不含目标物的空白儿童配方奶粉中,充分混匀,得到维生素K1和维生素K2含量均为75 μg/100 g的自制标准样品。乳粉中富含蛋白质和脂肪,维生素K1和维生素K2是脂溶性维生素,被包裹在乳粉颗粒中,不易直接提取,需要酶解、皂化,将其释放,经正己烷提取,浓缩复溶检测。
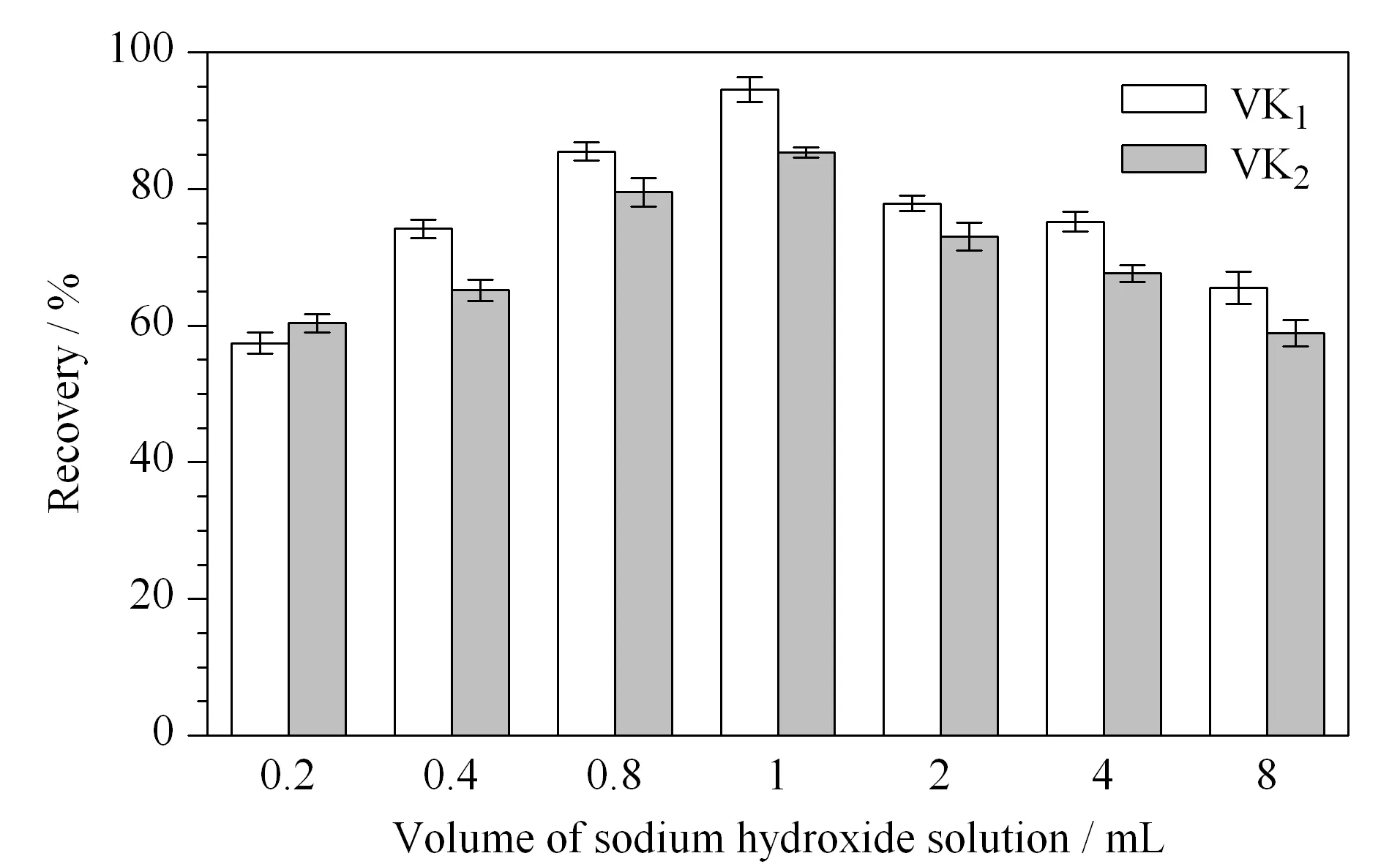
图 4 氢氧化钠溶液(2.5 mol/L)的加入量对维生素K1和维生素K2回收率的影响(n=3)Fig. 4 Effect of volumes of sodium hydroxide solution (2.5 mol/L) on the recoveries of the VK1 and VK2(n=3)
2.2.1 酶解条件的优化
酶解是调制乳粉中维生素K提取过程的重要环节之一,脂肪酶可将调制乳粉中的脂质降解成脂肪酸和甘油三酯。本文通过考察脂肪酶用量和酶解时间,对调制乳粉中维生素K1和维生素K2的提取效率进行考察。
脂肪酶的用量直接影响提取效率,不同酶解条件对维生素K1和维生素K2的影响趋势基本一致。实验考察了酶解过夜的条件下,不同脂肪酶的使用量,由图3a可见,脂肪酶的添加量为0.8 g时,维生素K1和维生素K2的回收率最高,分别为93%和91% 。脂肪酶用量不足,萃取时乳化严重,影响提取效率;脂肪酶使用过量会降低维生素K的提取效率,可能原因是酶使用量太高不利于乳粉的溶解,导致回收率下降。
当酶的添加量为0.8 g时,实验对不同酶解时间进行了考察。由图3b可见,酶解时间过短,酶解不完全,影响后续提取效率。酶解时间4 h以上,提取效率趋于稳定,回收率均达到90%以上。实验最终选择0.8 g的脂肪酶酶解4 h。

图 3 (a)脂肪酶用量和(b)酶解时间对维生素K1和维生素K2回收率的影响(n=3)Fig. 3 Effect of (a) dosage of lipase and (b) enzymolysis time on the recoveries of the VK1 and VK2(n=3)
2.2.2 皂化条件的优化
乳粉中维生素K经酶解后,通过皂化将脂肪酸变成水溶性脂肪酸钠,使脂溶性维生素K游离出来[22]。皂化条件,包括碱的加入量、皂化温度与皂化时间等,直接影响检测结果的准确性。如图4所示,本文对皂化条件中碱液(2.5 mol/L 的氢氧化钠溶液)的使用量进行了考察。结果表明,氢氧化钠溶液加入量为1 mL时的提取效率较好,维生素K1和维生素K2的回收率分别达到93%和92% 。氢氧化钠溶液加入量过少时,皂化不完全,液液萃取时不容易分层;加入量过多时,维生素K被碱破坏,均会影响提取效率。碱加入后,涡旋30 s后再加入10 mL无水乙醇,乙醇可以保护维生素K,还可以起到消泡作用和促进分层的作用。故本实验在酶解后的样液中加入1 mL 2.5 mol/L 氢氧化钠溶液,涡旋30 s后,再加入10 mL无水乙醇进行皂化。
2.2.3 萃取条件的优化
脂溶性维生素需采用有机溶剂萃取法进行提取,本文使用有机溶剂正己烷通过液液分配将维生素K1和维生素K2萃取出来;维生素K1和维生素K2在碱性条件下不稳定,水洗可以把萃取溶剂正己烷中微溶的碱液洗掉,选用饱和氯化钠溶液作为水洗溶液,即可防止液液分配过程的乳化,更有利于分层。本文同时对正己烷的用量(10 mL和15 mL)和萃取次数(1次和2次)进行了考察(见表1)。实验发现,采用正己烷10 mL或15 mL萃取2次,结果差异不大,对维生素K1和维生素K2的提取回收率均达到90%以上,但2次萃取对维生素K1和维生素K2的提取更充分。因此,从提高前处理效率和成本的角度,采用萃取条件为10 mL正己烷萃取2次。

表 1 萃取条件对维生素K1和维生素K2提取回收率的影响(n=3)Table 1 Effect of extraction conditions on the extraction recoveries of the VK1 and VK2(n=3)
2.3 方法学评价
2.3.1 线性范围和检出限
将浓度分别为 0.002 5、0.01、0.05、0.1、0.5、1.0和2.0 μg/mL 的维生素K1和维生素K2混合标准工作液注入液相色谱仪,按1.4节的液相色谱条件进行测定,以峰面积(y)为纵坐标,对应的质量浓度(x, μg/mL)为横坐标,绘制标准曲线,以3倍和10倍信噪比(S/N)确定检出限(LOD)和定量限(LOQ)。
结果表明,维生素K1在 0.002 5~2.0 μg/mL 范围内线性关系良好,线性相关系数(R2)>0.999,线性回归方程为y=2.37×106x-2.18×103, LOD和LOQ分别为0.07 μg/100 g和0.2 μg/100 g;维生素K2在0.01~2.0 μg/mL 范围内线性关系良好,R2>0.999,线性回归方程为y=1.55×106x-1.15×104, LOD和LOQ分别为0.24 μg/100 g和0.8 μg/100 g。
2.3.2 加标回收率和精密度
分别以低(5 μg/100 g)、中(50 μg/100 g)、高(100 μg/100 g)3个水平向空白样品中进行标准添加试验,每个水平平行测定6次,考察方法的回收率与精密度,结果见表2。方法的平均回收率为80.39% ~94.39% ,精密度为0.85% ~3.98% ,表明方法的精密度和准确度良好。
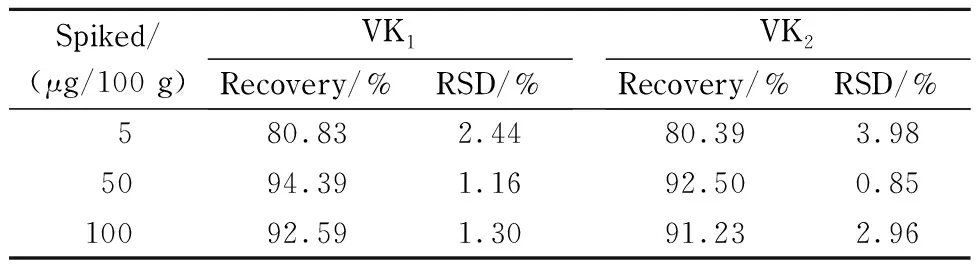
表 2 方法的回收率和精密度(n=6)Table 2 Recoveries and RSDs for the method (n=6)
2.4 实际样品检测
按照实验选择的最优条件对市售的10份儿童乳粉和10份孕产妇乳粉进行分析。检测结果显示:其中16份样品检出维生素K1,含量范围在35~70 μg/100 g之间,符合标示值及国家标准的规定值,其余4份未检出;维生素K2均未检出,推测原因是调制乳粉中维生素K添加剂仅为维生素K1,而维生素K2作为营养强化剂处于发展阶段,未在市售的调制乳粉中添加。
3 结论
本文对维生素K1和维生素K2的色谱条件和前处理条件进行优化,建立了柱后还原-高效液相色谱同时测定调制乳粉中维生素K1和维生素K2的分析方法。本方法定性定量准确,灵敏度高,重复性好,能满足调制乳粉中维生素K含量的检测要求,可以对维生素K1和维生素K2的质量控制和市场监管提供技术支持,有利于提高婴幼儿调制乳粉的安全性。
猜你喜欢
石油炼制与化工(2022年2期)2022-02-15
食品安全导刊(2021年21期)2021-08-30
化工管理(2020年26期)2020-10-09
中国乳业(2020年12期)2020-04-12
山东化工(2019年2期)2019-02-21
中国洗涤用品工业(2017年2期)2017-04-16
电子技术与软件工程(2016年24期)2017-02-23
天然产物研究与开发(2016年1期)2016-06-05
中国塑料(2016年7期)2016-04-16
食品安全导刊(2014年8期)2014-10-21
