
水分在托贝莫来石中运动特性的分子动力学模拟研究
2020-05-18 13:06:44杨铁军魏金龙马冰洋侯东帅
硅酸盐通报 2020年4期
杨铁军,魏金龙,马冰洋,侯东帅
(1.上海宝冶工程技术有限公司新型建筑材料厂,上海 201900;2.青岛理工大学土木工程学院,青岛 266033)
0 引 言
水泥基材料由于其低廉的价格和优异的力学性能被广泛应用于建筑和基础设施建设。但许多海洋工程或水中用的水泥基材料在使用一二十年或者更短的一段时间就会出现开裂和剥落等问题,导致结构的失效,从而造成巨大的经济损失。因此近些年来水泥基材料的耐久性越来越被人们所关注[1-2]。水化硅酸钙凝胶(Calcium Silicate Hydrate,C-S-H)是最主要的水泥水化产物, 占所有水泥水化产物总量的70%以上,水泥水化产物的性能很大程度上取决于C-S-H凝胶的性能。C-S-H凝胶是一种层状的多孔复杂结构,没有特定组成和固定的晶型,其内部的水分还会随温湿度的变化而改变[3-5]。更为关键的是水分还会携带氯离子和其他有害离子进入水泥基材料内部,导致基体开裂和内部的钢筋锈蚀等问题的发生[6-7]。
实验上通过各种技术研究了凝胶孔内部以及C-S-H表面水的性质,例如张鹏等[8-9]通过中子成像技术实现了混凝土中水分侵入过程的可视化追踪以及水分空间分布的定量计算。混凝土内部水分的扩散系数以及滞留时间等动力学参数则可以通过宽频介电谱试验(BDS)、中子散射试验(NS)等手段测得[10-11]。宏观实验对于混凝土内部水分传输的研究是不可或缺的,但仅采用宏观实验很难了解水分的微观传输机理。分子动力学模拟(MD)能对固-液界面的结构、动力学和能量进行更定量和详细的研究[12-13],了解物质在分子水平上的结构和动力学性质,帮助解释宏观实验现象。
本文采用分子动力学方法研究了托贝莫来石中(托贝莫来石11 Å和托贝莫来石14 Å,层间距不同) 水分子的结构和动力学特性,并通过均方位移(Mean Squared Displacement,MSD)、径向分布函数(Radial Distribution Function,RDF) 和氢键网络等参数进行表征。
1 模拟方法
1.1 模型构建
C-S-H凝胶是一种无定形的物质,这对于模型的构建十分不利。研究表明,C-S-H凝胶的结构与托贝莫来石结构十分相近,因此众多学者都采用托贝莫来石模型代替C-S-H凝胶来进行相关的模拟工作[14-15]。本次模拟采用的是托贝莫来石 11 Å和托贝莫来石 14 Å模型[16-17],溶液为0.5 mol/L、1.26 mol/L 和2.0 mol/L的CaCl2溶液。如图1(a)所示,CaCl2模型的尺寸为a=20.69 Å,b=20.69 Å,c=20.69 Å,α=90°,β=90°,γ=90°,0.5 mol/L 的CaCl2溶液模型包含300个水分子,2个Ca2+和4个Cl-(1.26 mol/L的CaCl2溶液包括5个Ca2+和10个Cl-,2.0 mol/L的溶液包括8个Ca2+和16个Cl-),溶液的密度都在1 g/cm3左右。托贝莫来石11 Å模型的晶胞尺寸为a=22.32 Å,b=22.17 Å,c=22.77 Å,α=90°,β=90°,γ=90°,见图1(b)。托贝莫来石14 Å模型的晶胞尺寸为a=22.53 Å,b=22.28 Å,c=27.99 Å,α=90°,β=90°,γ=90°,见图1(c)。
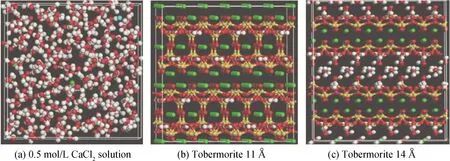
图1 初始模型示意图(图(a)中球棍代表水分子,单个球体代表离子;图(b)(c)中链状结构代表硅链,球棍代表水分子,单个球体代表钙离子)Fig.1 Schematic of the initial model(in fig.(a) the ball-stick represents water molecule, the single ball represents ion;in fig.(b), (c), the ball-chain represents the silicon chain, the ball-stick represents the water molecule, and the single ball represents the calcium ion)
1.2 力场与模拟流程
模拟采用CLAYFF力场[18],该力场已经广泛应用于溶液/氢氧化物界面的相互作用,不同水泥相(包括托贝莫来石,AFm和蒙脱石)层间结构中水和离子行为的研究[19]。
计算工作在Lammps软件中完成,模拟采用NVT系综(原子数量、体积、温度保持不变),体系温度设置为298 K,步长为1 fs,总共持续100 ps,输出的平衡动力学轨迹一共包含100 000帧的原子构型。足够大的样本数据可以确保数据分析的统计稳定性。
1.3 数据分析
径向分布函数(RDF)、均方位移(MSD)和氢键网络是评估不同模型中水分子结构和动力学性质中常用的参数。RDF可用于分析系统的结构信息,表示结构的有序程度,如式(1)所示。
g(r)=dN/(4πr2ρ)
(1)
RDF原理图如图2所示,公式(1)中dN代表dr范围内的原子数;ρ表示系统的平均密度。该式可以解释为系统的区域密度和平均密度之比,通过分析RDF峰值的尖锐程度、峰宽以及相对位置,可以得到离子和离子、离子和固体原子以及水分子和固体氧原子的空间相关性。
均方位移(MSD)则是用于评估原子动力学特性的参数,具体公式如式(2)所示。
MSD=〈|ri(t)-ri(0)|2〉
(2)
式中,ri(t)代表i原子在t时刻的位置,ri(0)代表i原子在初始时刻的位置。MSD越大,代表原子距离起始位置越远,表明运动越剧烈。

图2 RDF原理图Fig.2 Schematic for RDF
由于托贝莫来石表面具有亲水性,氢键广泛存在于托贝莫来石和水分子的之间,因此氢键网络对于表征水分子构型以及托贝莫来石与表面水分子之间的相互作用起着至关重要的作用。
氢键网络结构由水分子和托贝莫来石表面的氢氧元素构成。为了描述局部氢键环境,有必要分别计算每个水分子贡献给相邻水分子和贡献给托贝莫来石表面的平均氢键数。氢键的形成需要满足两个条件,如图3所示,(1)两个相邻水分子DH-O之间的距离应小于2.45 Å。(2)OH向量和OO向量之间的角θ应小于30°[20]。为了便于理解,将提供氢原子的水分子称为供体水分子,提供氧原子的水分子成为受体水分子,图3中左上方的水分子为贡体水分子,右下方为受体水分子。
2 结果与讨论
2.1 径向分布函数
图4为三个模型Ow-Hw、Ow-Ow、Hw-Hw的RDF曲线,其中分子内的RDF峰值已被忽略。RDF曲线为系统的区域密度和平均密度之比,可以简单理解为峰越明显,则相应横坐标处的原子数量越多,空间相关性越强。从图4(a)中可以看到CaCl2溶液中Ow-Hw的RDF曲线仅在1.8 Å和3.2 Å处存在峰值,1.8 Å处的峰值为氢键的特征峰,3.2 Å处的峰值则为氢键的次特征峰(即两个相邻氢键导致的空间相关性)。溶液中各原子对的RDF曲线仅在近程5 Å以内的地方存在峰值,超过5 Å后峰值便逐渐消失,这表明溶液中水分子之间的空间相关性较弱,水分子的移动几乎不受限制。
托贝莫来石11 Å模型的RDF曲线峰值分布与溶液水的表现完全不同,以Ow-Hw为例,除了在1.8 Å附近有氢键特征峰以外,还在4.7 Å、5.9 Å、7.6 Å等多处都存在峰值。并且与溶液水相比,11 Å模型在1.8 Å处的氢键特征峰明显弱化,3.2 Å处的峰值则基本消失,这意味着11 Å模型中水分子间的氢键连接大幅减少,氢键特征峰的变化以及远程特征峰的出现,表明水分子受到了托贝莫来石界面的强烈影响,表现出了与溶液水截然不同的空间分布。
接着来看14 Å模型的RDF曲线,可以发现其特征峰值分布与溶液水相似,同时在5 Å外还存在着一两个特征峰,这表明14 Å模型中水分子的空间分布与溶液水较为相似,但远程空间相关性略微强于溶液水。14 Å模型和11 Å模型的不同之处在于其层间距的增加,这说明层间距的变化对水分子空间分布有着重要的影响。这是因为随着层间距的增大,硅链对水分子的空间限制作用减弱,水分子可以较为自由的移动,导致其RDF曲线特征峰减少,逐渐向溶液水转变。同时还要注意,14 Å模型的RDF曲线的特征峰要强于溶液水,这说明托贝莫来石模型对水分子除了有空间限制效果外,还存在着另外的影响,由下面的分析可知,这种影响为界面对水分子的氢键吸附作用。从RDF曲线的分析中可以看出,被限制在托贝莫来石中的水比溶液中的水更有序,远距离的空间相关性增加。

图4 各原子对的RDF曲线(Ow和Hw分别代表水中的氧原子和氢原子)Fig.4 Radial distribution function (Ow and Hw refer to O atom and H atom in water molecule)
2.2 氢 键
托贝莫来石是一种亲水结构,其内部水分子会与结构形成氢键连接,这对水分子的迁移有着重要的影响,因此计算每个水分子周围的平均氢键数是十分必要的。图5(a)为CaCl2溶液中水分子的局部结构图,可以非常明显地看到CaCl2溶液中水分子与相邻水分子形成了氢键。从表1中可以了解到CaCl2溶液中每个水分子平均会和周围水分子形成1.6个贡献氢键和1.6个接受氢键,总计和周围水分子形成3.2个氢键。图5(b)为托贝莫来石11 Å模型中的水分子的局部结构图,可以看到托贝莫来石11 Å模型中的水分子不再与相邻水分子形成氢键,而是与托贝莫来石中的硅链形成氢键,从表1了解到托贝莫来石11 Å模型中平均每个水分子都会形成1个此类氢键,且没有其他种类的氢键形成。图5(c)中可以看出托贝莫来石14 Å模型中的水分子不仅保留着水分子之间的氢键同时还与硅链形成氢键,平均每个水分子可以与Os(二氧化硅四面体中的氧原子)形成0.64个氢键、与Ho(来自硅羟基中的氢原子)形成0.2个氢键、与周围水分子形成0.71个贡献氢键和1.1个接受氢键。
托贝莫来石模型与水分子都形成了一定数量的界面氢键,11 Å模型和14 Å模型的界面氢键数量分别为1、0.84(Hw-Os以及Ho-Ow都属于界面氢键)。由于界面氢键比水分子之间的氢键更为稳定,因此水在托贝莫来石模型中的运动将受到更多的限制[21]。
同时还要注意与11 Å模型相比,14 Å模型中水分子和硅链形成的氢键数量在减少,与相邻水分子形成的氢键数量则从0增加到了1.81,这表明14 Å模型和水分子的空间相关性较11 Å模型要弱,这与RDF分析的结果是一致的。

图5 水分子局部结构图(虚线表示氢键)Fig.5 Local structure diagram of water molecules
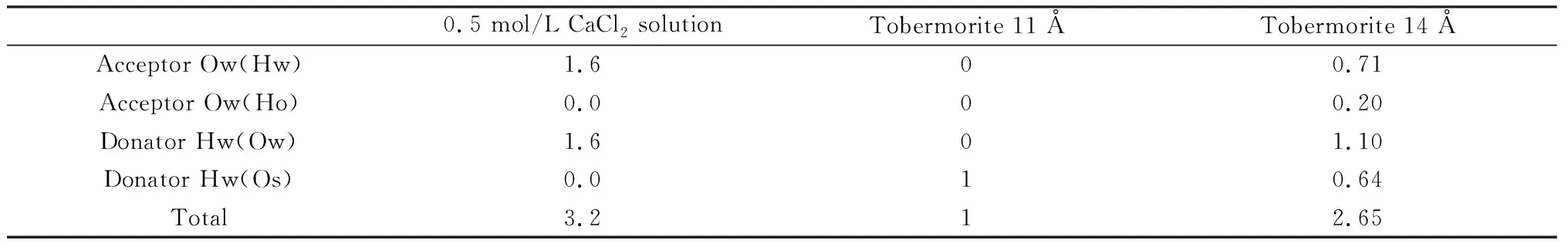
0.5 mol/L CaCl2 solutionTobermorite 11 ÅTobermorite 14 ÅAcceptor Ow(Hw)1.600.71Acceptor Ow(Ho)0.000.20Donator Hw(Ow)1.601.10Donator Hw(Os)0.010.64Total3.212.65

图6 水分子MSD曲线Fig.6 MSD curves of water molecules
2.3 均方位移
MSD曲线可以用来表征水分子的动力学特性,相同时间内MSD数值越大,代表原子距离起始位置越远,表明运动越剧烈。图6(a)为托贝莫来石模型的MSD曲线,14 Å模型的曲线在10 ps时达到了1 Å2,而11Å模型中这一数值仅为0.5 Å2,这说明11 Å模型中的水分子运动要明显弱于14 Å模型中的水分子,结合先前结构分析可知这是11 Å模型的层间距小,空间几何限制强烈以及11 Å模型与水分子形成了较多的界面氢键导致的结果。此外,托贝莫来石模型中水分子的MSD曲线在0.2 ps左右出现了一个小的平台,之后MSD曲线的斜率明显放缓,表现为典型的固态物质的运动特性曲线。
图6(b)为0.5 mol/L、1.26 mol/L、2.0 mol/L CaCl2溶液的MSD曲线,溶液水的MSD随时间的增加而增大,近似于一条直线,表现为典型的液体物质的运动特性。对比图6(a)和图6(b)可以发现,在相同的10 ps时间内,托贝莫来石中水分子的MSD数值(1 Å2以内)要比溶液水(10 Å2左右)小一个数量级以上。由之前的结构分析可知,由于界面氢键作用和托贝莫来石层的约束[22-23],使层间水分子不能像溶液中水一样自由移动,因此两者的运动速率相差较大。在图6(b)中还可以发现不同浓度溶液中水分子的运动速率为:0.5 mol/L>1.26 mol/L>2.0 mol/L,水分子的运动速度随着溶液浓度的增加而降低。这是因为在浓度高的溶液中水分子将会被更多离子(例如Ca2+和Cl-)吸引,使水分子的运动速度降低。
3 结 论
通过分子动力学模拟研究了托贝莫来石以及溶液中水分子的结构和动力学行为,得到以下结论:
(1)托贝莫来石11 Å模型中水分子受空间几何限制以及界面氢键的原因,在10 Å范围内表现出了强烈的空间相关性;托贝莫来石14 Å模型层间距增大,几何限制作用变弱,界面氢键数量减少,水分子间的空间相关性变小,但由于水分子和托贝莫来石表面形成了氢键,其空间相关性要强于溶液水分子。
(2)通过MSD曲线发现,托贝莫来石中水分子的运动远小于溶液水分子,两者10 ps时的MSD数值相差一个数量级以上。托贝莫来石11 Å模型中的水分子相较于托贝莫来石14 Å模型表现的更为稳定;溶液中水分子的运动速率随着离子浓度的增加而降低。
猜你喜欢
中学生理科应试(2024年1期)2024-05-18 13:02:52
科教新报(2021年11期)2021-05-12 19:50:11
陶瓷学报(2020年6期)2021-01-26 00:38:08
山东陶瓷(2020年5期)2020-03-19 01:35:28
陶瓷学报(2019年6期)2019-10-27 01:18:22
陶瓷学报(2019年6期)2019-10-27 01:18:20
科学之谜(2016年9期)2016-10-11 08:59:04
原子与分子物理学报(2014年3期)2014-02-28 22:18:23
无机化学学报(2014年1期)2014-02-28 17:30:06
少儿科学周刊·儿童版(2012年5期)2012-08-30 20:12:50
