
托匹司他的合成研究进展
2020-05-13 08:27:06卢文龙古双喜徐志强
合成化学 2020年4期
卢文龙, 陈 炯, 古双喜, 徐志强
(武汉工程大学 化工与制药学院 绿色化工过程教育部重点实验室,湖北 武汉 430205)
高尿酸血症是由各种遗传和环境因素引起血尿酸(UA)代谢紊乱而导致的疾病[1],具体表现为血清中尿酸盐水平异常升高,超过生理温度和pH下的溶解度极限(约6.8 mg/dL),在关节毛细血管中会形成微晶,从而导致痛风[2]。在过去50年中,世界各个地区痛风患病率日益增长,对人们的健康造成了巨大的威胁[3]。目前在临床上,药物主要通过抑制UA的生成对痛风进行治疗,降低黄嘌呤氧化酶(XOR)的活性,抑制次黄嘌呤转化成黄嘌呤,进而减少UA的生成,降低血液中UA的浓度。代表药物主要有别嘌呤醇、非布司他、托匹司他(Chart 1)。
托匹司他(Topiroxostat,1),化学名为5-(2-氰基-4-吡啶基)-3-(4-吡啶基)-1,2,4-三唑[4],由日本富士药品株式会社研发,并于2013年在日本获批上市,但该药物在国内还未上市[5]。作为一种新型的XOR抑制剂,与别嘌呤醇相比,1在体内和体外均表现出更强的活性,但在体内的作用时间更长[6]。1对还原和氧化型的XOR均有抑制作用,药效好,不良反应小,安全性强,是目前治疗高尿酸血症的有效的药物之一。
Scheme 1
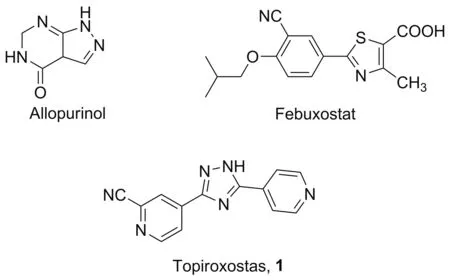
Chart 1
在结构上,1由三唑环片段和两个吡啶环片段拼合而成,其合成关键步骤在于吡啶2位氰基的引入(Ⅰ)、两个吡啶环的拼接(Ⅱ)和三唑环的构建(Ⅲ)。根据各关键步骤的顺序不同,将托匹司他的合成策略分为以下3种:“Ⅰ+Ⅱ+Ⅲ”、“Ⅱ+Ⅲ+Ⅰ”和“Ⅱ+Ⅰ+Ⅲ”。本文主要以这3种策略为主线介绍1自2002年发现至今的合成研究进展,旨在为开发更适合工业化生产的路线提供参考。
1 Ⅰ+Ⅱ+Ⅲ合成1
文献报道1的合成路线大部分是通过Ⅰ+Ⅱ+Ⅲ合成策略实现的,即:首先在吡啶2-位引入氰基(Ⅰ),再构建三唑环(吡啶环的拼接和三唑环的构建可以一锅完成,Ⅱ+Ⅲ)。有些工艺路线使用吡啶2位含有氰基的化合物为起始原料,该类路线也被归为策略Ⅰ+Ⅱ+Ⅲ中。起始原料分为以下几种:异烟酸-N-氧化物(2);2-氰基异烟酸(10);异烟酸甲酯(12);异烟酸甲酯-N-氧化物(3);2-氰基异烟酸甲酯(4); 2-氯异烟酸(16)。
1.1 以异烟酸-N-氧化物(2)为原料
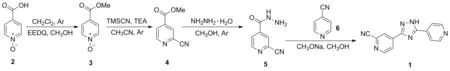
2002年日本富士药品株式会社申请了以2为起始原料合成1的专利[7],报道了1的第一条合成路线(Scheme 1)。原料2与1-乙氧基羰基-2-乙氧基-1,2-二氢喹啉(EEDQ)在氩气保护下酯化得到异烟酸甲酯-N-氧化物(3),收率为72.6%;在氩气保护下,于乙腈中,3与三甲基氰硅烷(TMSCN)在三乙胺(TEA)的作用下反应得到2-氰基异烟酸甲酯(4),收率为71.8%;在氩气保护下,4在甲醇中与水合肼反应,得到2-氰基异烟肼(5),收率为49.2%; 5与4-氰基吡啶(6)在甲醇钠/甲醇体系中发生关环反应得到产物1,收率为57.6%。
该路线总收率仅为14.8%,适于小批量制备目标化合物。该路线起始原料2和TMSCN价格昂贵(后者有剧毒),前两步反应时间过长,且需要柱层析,因而难以应用于工业化生产。此外,4与水合肼发生缩合反应的同时,吡啶2-位的氰基也会与水合肼发生副反应,生成杂质A,该杂质也可以与4反应生成杂质B(Chart 2),其溶解度较差,在后处理过程中难以除去,对原料药的质量影响较大。
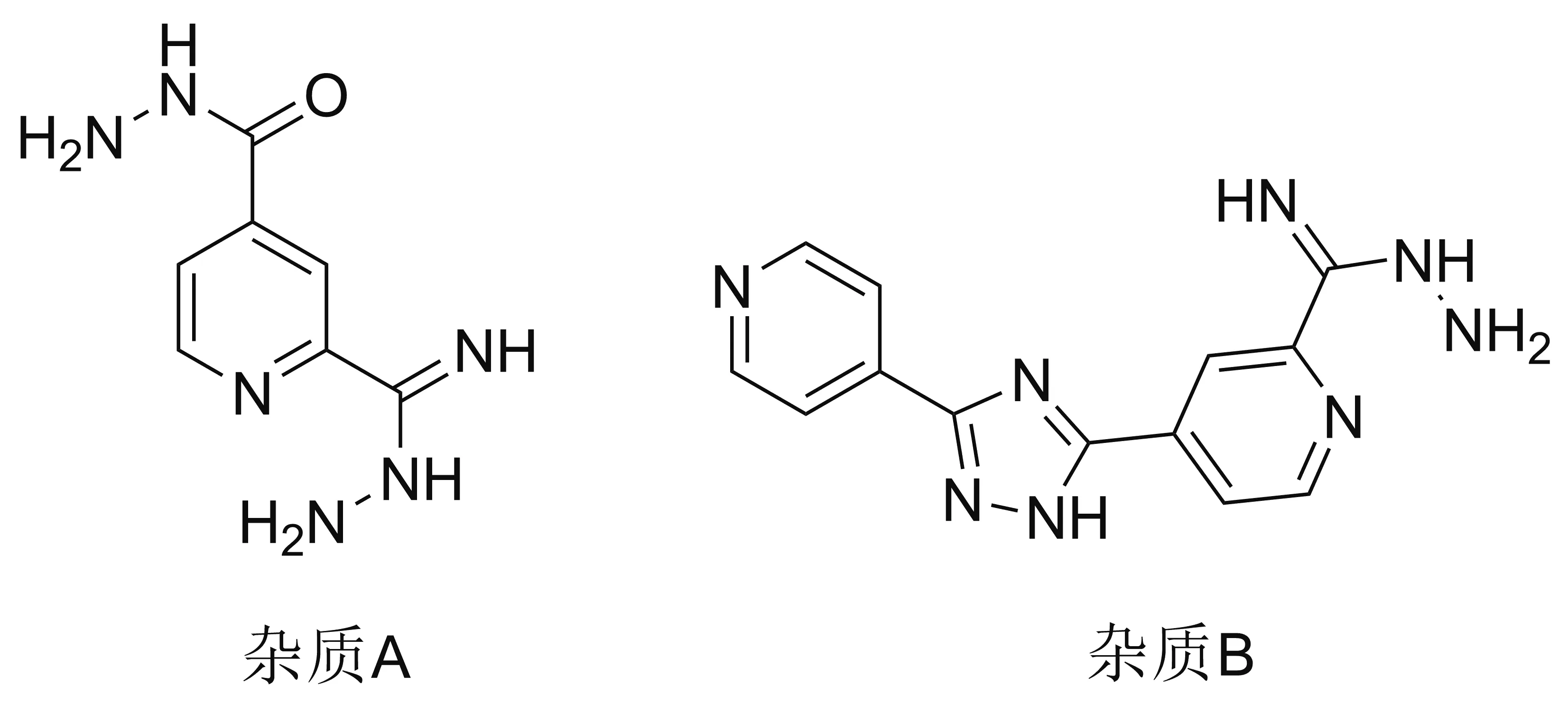
Chart 2
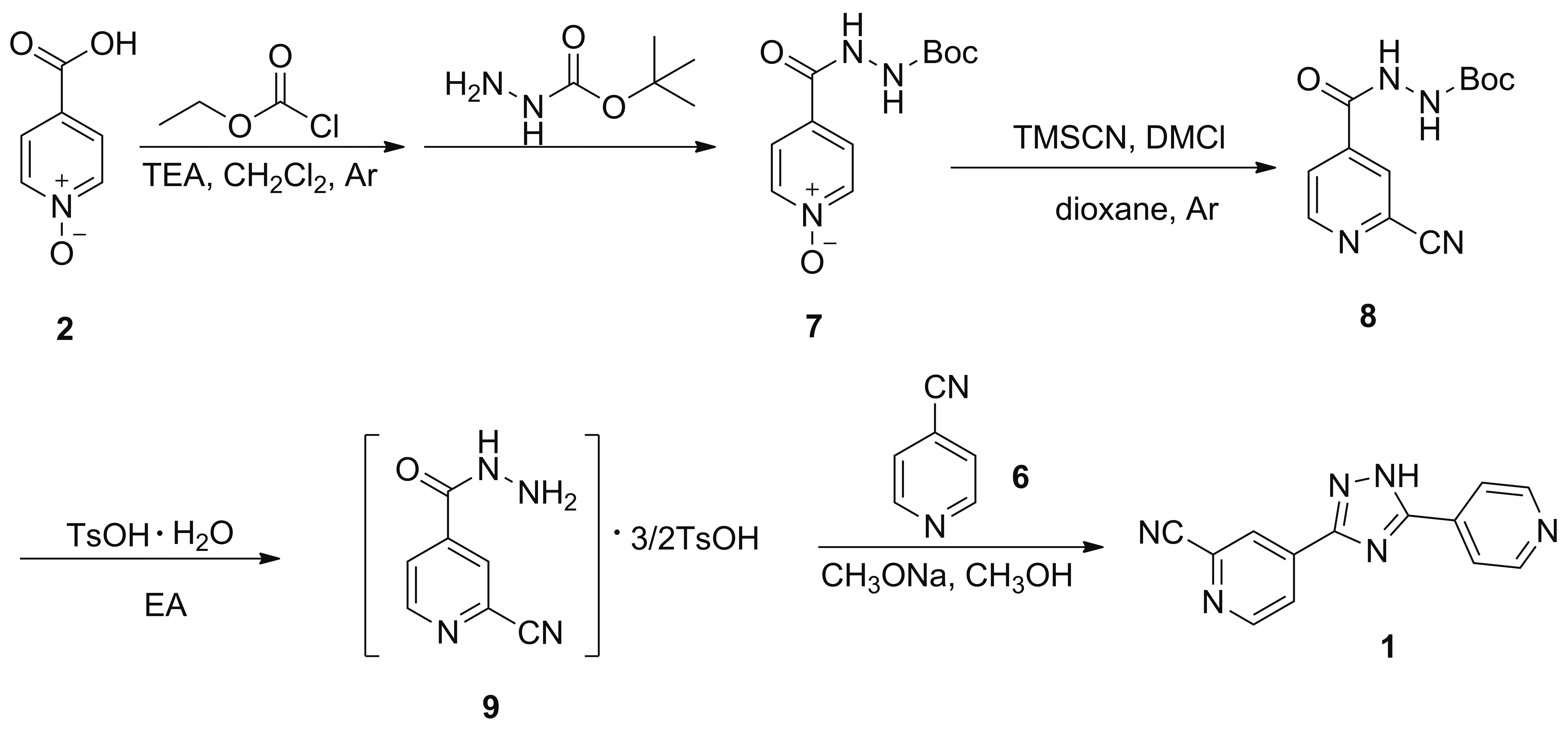
为避免该副反应的发生,该专利还报道了另一条路线(Scheme 2)[7],该方法于2008年被霍志宝等[8]用于1的工艺改进。原料2与氯碳酸乙酯,于搅拌下加入肼甲酸叔丁酯反应得到4-(2-(叔丁氧羰基)肼基-1-羰基)吡啶-N-氧化物(7),收率为70.0%; 7在TMSCN和N,N-二甲氨基甲酰氯(DMCl)的作用下于氮气保护下在二氧六环中发生氰化反应得到2-氰基-4-(2-(叔丁氧羰基)肼基-1-羰基)吡啶(8); 8与对甲苯磺酸一水合物于乙酸乙酯(EA)中反应得到2-氰基异烟肼·3/2对甲苯磺酸盐(9),两步收率为79.9%; 9与6在甲醇钠/甲醇体系中发生关环反应得到产物1,收率为68.4%[7]。该路线相比于Scheme 1,分离纯化较为简单,使用合适的溶剂打浆即可获得纯度较高的产物。同时避免了氰基与水合肼的副反应。但肼位引入叔丁氧羰基,增大了位阻,导致缩合反应转化率低,且反应速度慢,收率低,导致成本较高,并且工序复杂,不适合工业化生产。
Scheme 2
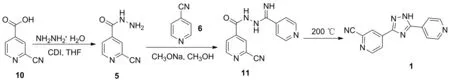
Scheme 3
1.2 以2-氰基异烟酸(10)为原料
2009年,佐藤隆宽等报道了以10为起始原料合成1的工艺路线(Scheme 3)[6,9]。于四氢呋喃(THF)中,10在N,N′-羰基二咪唑(CDI)作用下与水合肼发生肼解反应得到5; 5与6在甲醇钠/甲醇体系中缩合制得2-氰基-N-(亚胺基-(4-吡啶基)甲基)异烟酰肼11; 11在200 ℃下闭环得到产物1。该路线前两步反应后处理经过打浆即可分离纯化,但第三步反应(高温关环)并不理想,事实上,这一关环反应可与第二步反应一锅发生生成1[5,7,10,12]。此外,该路线的起始原料10价格昂贵,不具备成本优势,不适合工业化生产。
1.3 以异烟酸甲酯(12)为原料
2013年,孙元军等[10]报道了以12为起始原料,用酰胺脱水的方法在吡啶2-位引入氰基的合成路线(Scheme 4)。原料12在甲酰胺、浓硫酸、七水硫酸亚铁和30%双氧水的作用下发生酰胺化反应制得2-氨基甲酰基异烟酸甲酯(13),收率为38.7%;在二甲基甲酰胺(DMF)中,13经三聚氯氰脱水制得4,收率为87.7%; 4在甲醇中与水合肼发生肼解反应得到5,收率为50.0%; 5与6在甲醇钠/甲醇体系中发生关环反应得到产物1,收率为69.1%。该路线所用试剂价格低廉,降低了成本,同时避免使用剧毒氰化物,用酰胺脱水的策略引入氰基,方法新颖,安全性高,但收率低,四步反应总收率仅为11.7%,且工序繁琐(第一步反应中七水硫酸亚铁和30%双氧水须分多次交替加入,对设备要求高,且后处理过程较为繁琐),不适合工业化生产。
2014年,张梦甜报道了以12为起始原料,经过氧化,氰化,肼解,成环制得1的工艺路线(Scheme 5)[5]。原料12在间氯过氧苯甲酸(m-CPBA)作用下于二氯甲烷(DCM)中发生氧化反应制得3[11],收率为98.5%; 3与TMSCN在TEA的作用下于甲醇中发生氰化反应得到4,收率为65.8%; 4在甲醇中与水合肼发生肼解反应得到5,收率为49.2%; 5与6在甲醇钠/甲醇体系中发生关环反应得到产物1,收率为57.6%。该路线使用的原料和试剂价格便宜,具有一定的成本优势,反应条件温和,操作简便,但路线总收率较低(仅为18.4%),还需进一步优化以提高收率,才能具有工业化生产的竞争力。
2015年,张颖等[12]报道了以12为起始原料,在三氯氧磷(POCl3)、 DMF、碘和28%氨水条件下,一锅法在吡啶2-位引入氰基的工艺路线(Scheme 6)。原料12首先在POCl3和DMF的条件下,形成过渡态,然后在I2和28%氨水的条件下发生氰化反应得到4,收率为41.2%; 4在甲醇中与水合肼发生肼解反应得到5,收率为48.1%; 5与6在甲醇钠/甲醇体系中发生关环反应得到粗品1,收率为60.5%;粗品1在水/2-丁醇体系中与对甲苯磺酸一水合物反应得到托匹司他对甲苯磺酸盐(14),收率为87.7%; 14在乙醇水溶液中与碳酸氢钠反应得到1,收率为90.3%。该路线与Scheme 4方案相比,仅第一步引入氰基的方法不同。用POCl3/DMF/I2/氨水反应体系引入氰基,避免使用剧毒氰化物,后处理操作简单,经过重结晶即可得到较纯的产物,但此步骤收率太低,导致该路线总收率仅有9.5%。同时,该路线使用对甲苯磺酸一水合物精制,会产生基因毒性杂质(对甲苯磺酸酯,Chart 3)。综上,该法适合高纯度1的小批量生产,不适合工业化。
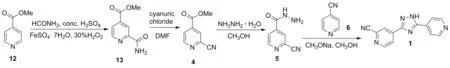
Scheme 4
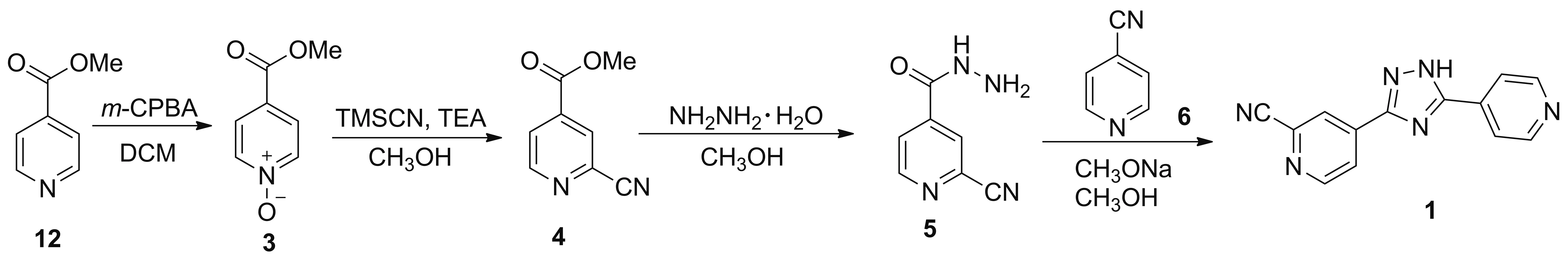
Scheme 5
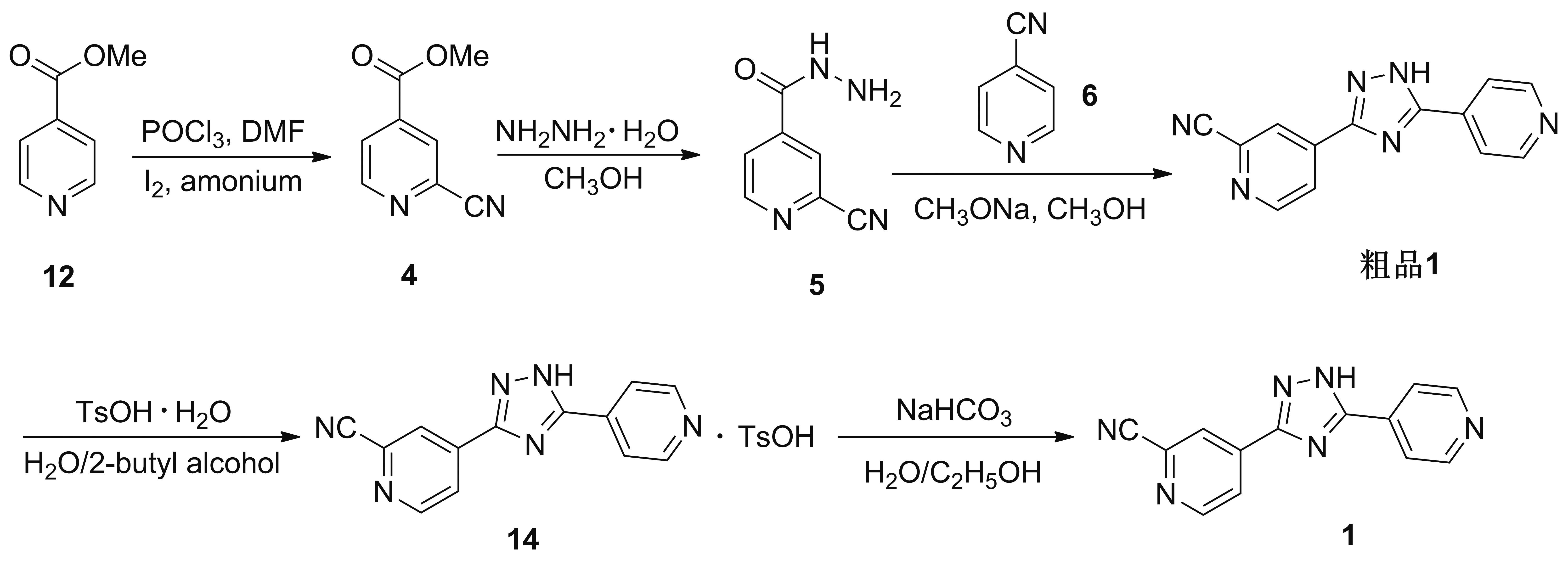
Scheme 6

Chart 3
2017年,王立新等[13]报道了以12为起始原料制备1的工艺路线,同时也提供了一种环保、适合工业化的晶型制备方法(Scheme 7)。原料12在甲酰胺、浓硫酸、双氧水和硫酸铁条件下发生酰胺化反应制得13,收率为76.7%;于EA中,13在三氟乙酸酐(TFAA)和二异丙基乙胺(DIEA)条件下脱水得到4,收率为84.9%; 4在甲醇中与水合肼发生肼解反应制得5; 5与6在甲醇钠/甲醇体系中缩合制得11,两步收率为90.0%; 11在乙醇钠/乙醇体系中发生关环反应得到粗品1,收率为92.0%;粗品1在乙醇水溶液中与氢溴酸成盐制得托匹司他氢溴酸盐(15),收率为90.5%; 15在碳酸钠的异丙醇(IPA)/水溶液中反应制得1,收率为93.9%。该路线与Scheme 4类似,同样使用酰胺脱水的策略引入氰基,不足之处也与其相同;但在精制时使用氢溴酸成盐,比对甲苯磺酸更有优势,避免了对甲苯磺酸酯杂质的产生,为1的精制提供了新的思路。
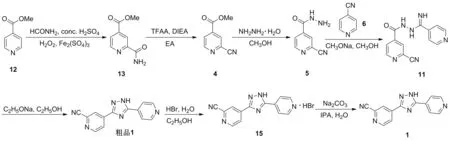
Scheme 7
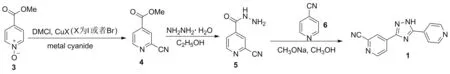
Scheme 8
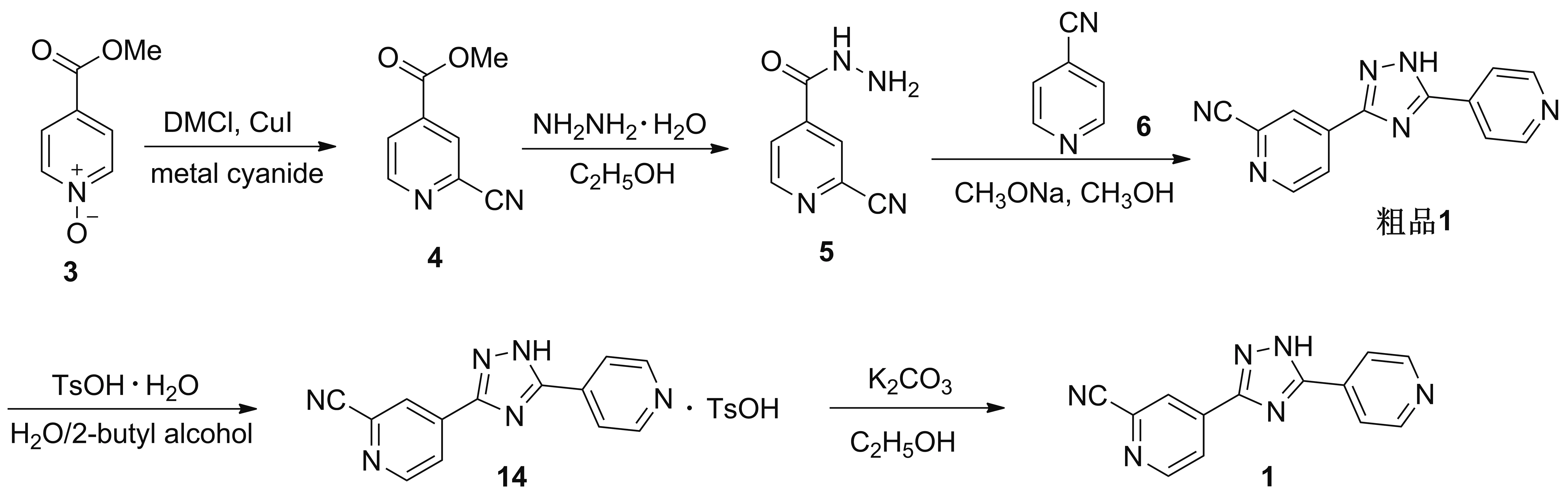
Scheme 9
1.4 以异烟酸甲酯-N-氧化物(3)为原料
2014年,王庆本等[14]报道了以3为起始原料合成1的工艺路线(Scheme 8)。原料3在铜催化剂(10% eq.)、金属氰化物(1.2 eq.)和DMCl(1.1 eq.)作用下发生氰化反应得到4,收率为94%; 4在乙醇中与水合肼发生肼解反应得到5,收率为83%; 5与6在甲醇钠/甲醇体系中发生关环反应得到产物1,收率为78%。该路线较简短,总收率在61%以上,但后处理操作繁琐,需要柱层析;使用金属氰化物引入氰基,相比酰胺脱水的方法,安全性较低;且起始原料3价格昂贵,成本较高,不适合工业化生产。
高瑞等[15]于2016年报道了以3为起始原料合成1的工艺路线(Scheme 9)。该路线前三步与Scheme 8方法相同,3步反应的收率依次为92.0%、 86.0%和63.0%。区别在于该路线使用对甲苯磺酸一水合物水溶液/2-丁醇体系精制粗品1,制得14,收率为95%; 14在碳酸钾的乙醇溶液中反应得到1,收率为98.0%。该法优于Scheme 8的是后处理纯化采用重结晶的方法,避免了柱层析,操作简单,得到的产品1纯度较高(98%)。但都没有避免使用金属氰化物,安全性较低;同时,对甲苯磺酸一水合物会与醇发生副反应产生基因毒性杂质对甲苯磺酸酯,这是该路线存在的另一问题。
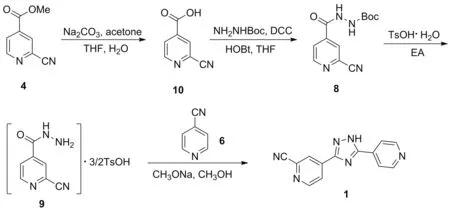
Scheme 10
Scheme 11
Scheme 12
1.5 以2-氰基异烟酸甲酯(4)为原料
2015年,陈云等[16]报道了以4为起始原料合成1的工艺路线(Scheme 10)。原料4在碳酸钠的作用下于丙酮/四氢呋喃/水混合溶剂中反应得到10,收率为86%; 10在N,N′-二环己基碳酰亚胺(DCC)、1-羟基苯并三唑(HOBt)的作用下,于THF中与Boc-肼发生肼解反应得到8,收率为87%; 8与对甲苯磺酸一水合物于EA中成盐制得9,收率为74%; 9与6在甲醇钠/甲醇体系中发生关环反应得到产物1,收率为57%。该路线总收率仅为32%,起始原料4价格昂贵,因目前市售的中间体10比起始原料4更廉价,故第一步反应并无实际意义,不适合工业化生产。
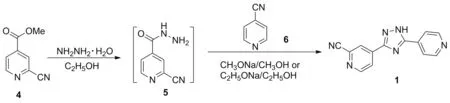
2016年,王德才等[17]报道了以4为起始原料的一锅法合成工艺(Scheme 11)。原料4在乙醇中与水合肼发生肼解反应得到5; 5与6在甲醇钠/甲醇体系中发生关环反应制得产物1。总收率为62.1%。2017年刘兴超等[18]也报道了该路线的分步合成法,原料4在乙醇中与水合肼发生肼解反应得到5;收率为85.0%;5与6在乙醇钠/乙醇体系中发生关环反应得到产物1,收率为87.1%。总收率在62%以上。该路线相比于其他方案,较为简短,总收率也较高,但起始原料4价格昂贵,如能开发其简易合成法,该路线将是一条极具竞争力的工艺路线。
Scheme 13
1.6 以2-氯异烟酸(16)为原料
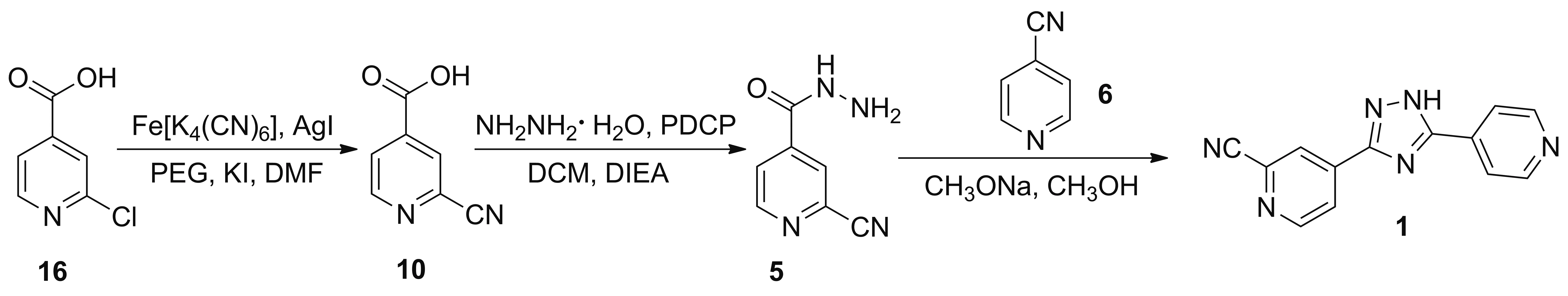
2016年,孙松等[19]报道了以16为起始原料合成1的工艺路线(Scheme 12)。在DMF中,原料16于亚铁氰化钾(0.2 eq.)、碘化银(3% eq.)、碘化钾(3% eq.)和聚乙二醇(4% eq.)的作用下发生氰化反应制得10,收率为84.5%; 10在二氯化磷酸苯酯(PDCP)和DIEA的作用下于DCM中与水合肼发生肼解反应制得5,收率为92.3%; 5与6在甲醇钠/甲醇体系中发生关环反应得到产物1,收率为85.6%。该路线相比于其他方案,是目前最优的合成工艺路线:其步骤少,收率高,总收率在66.8%以上;且分离纯化使用重结晶的方法,操作简单,反应时间短,条件温和;原料16廉价易得,工艺成本低;相比于酰胺脱水的方法引入氰基,使用亚铁氰化钾作为绿色氰源,其收率更高,同时避免使用价格高昂的剧毒氰化物,经济环保。
该策略主要是先构建三唑环(吡啶环的拼接和三唑环的构建可以一锅完成)(Ⅱ+Ⅲ),然后在吡啶2-位引入氰基(Ⅰ)。起始原料分为以下几种:异烟肼(17); 4-氰基吡啶-N-氧化物(18);异烟酸甲酯-N-氧化物(3); 4-氰基吡啶(6)。
2.1 以异烟肼(17)为原料
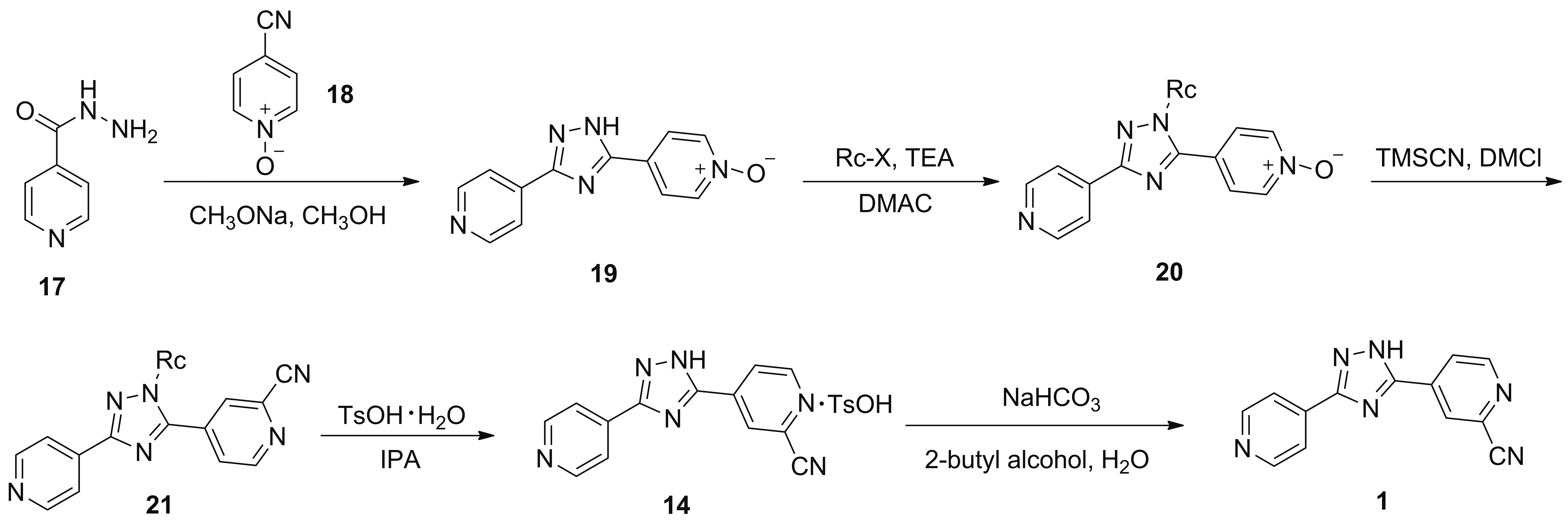
2004年,富士药品株式会社[20]报道了以17为起始原料合成1的工艺路线(Scheme 13)。17与4-氰基吡啶-N-氧化物(18)在甲醇钠/甲醇体系中发生关环反应得到产物4-(3-(4-吡啶基)-1H-1,2,4-三唑-5-基)吡啶1-氧化物(19),收率为90.5%;于N,N-′二甲基乙二胺(DMAC)中,19在苄基氯甲基醚和TEA的作用下,在三唑环NH上引入保护基得到N-苄氧甲基-5-(1-氧-4-吡啶基)-3-(4-吡啶基)-1,2,4-三唑(20); 20在TMSCN和DMCl作用下发生氰化反应得到N-苄氧甲基-5-(2-氰基-4-吡啶基)-3-(4-吡啶基)-1,2,4-三唑(21); 21在IPA中与对甲苯磺酸一水合物反应得到14,三步收率为61.5%; 14在2-丁醇/水的体系下与碳酸氢钠反应得到1,收率为99.6%。该路线步骤繁琐,中间体溶解性较差,需使用大量溶剂萃取洗涤,增加成本;且反应时间长,使用对甲苯磺酸一水合物精制,会产生杂质对甲苯磺酸酯;同时,使用剧毒氰化物引入氰基,环保压力大,不利于工业化生产。
2008年,霍志宝等[8]报道了以17为起始原料合成1的工艺路线(Scheme 14)。 17与18在甲醇钠/甲醇体系中发生关环反应得到产物19;于乙腈中,19在氰化锌和DMCl作用下发生氰化反应得到5-(2-氰基-4-吡啶基)-N,N-二甲基-3-(4-吡啶基)-1H-1,2,4-三唑-1-甲酰胺(22); 22与对甲苯磺酸在IPA和甲苯体系中反应得到14。该路线较简短,原料廉价易得;但氰化锌用量较大,且该步骤收率仅有66.0%,氰化锌同样有剧毒,相比TMSCN,并无优势;化合物19,溶解性较差,氰化锌在有机溶剂中同样难以溶解,底物无法与氰化试剂充分接触,需升高温度和延长反应时间;该工艺还使用对甲苯磺酸一水合物,其会与醇发生副反应产生基因毒性的杂质对甲苯磺酸酯,该路线同样不适合工业化生产。
2014年,庄妍[21]申请了以17为起始原料合成1的专利(Scheme 15), 17与18在甲醇钠/甲醇体系中发生关环反应得到产物19,收率为78%;于乙腈中,19在氰化锌(1.2 eq.)、 DMCl(2.2 eq.)和碘化亚铜(0.1 eq.)作用下发生氰化反应得到22收率为93.0%; 22与对甲苯磺酸在IPA中反应得到14,收率为100.0%; 14在乙醇水溶液中与碳酸氢钠反应得到产物1,收率为95.0%。该路线同样使用了氰化锌和对甲苯磺酸,缺点和Scheme 14相同,不适合工业化生产。
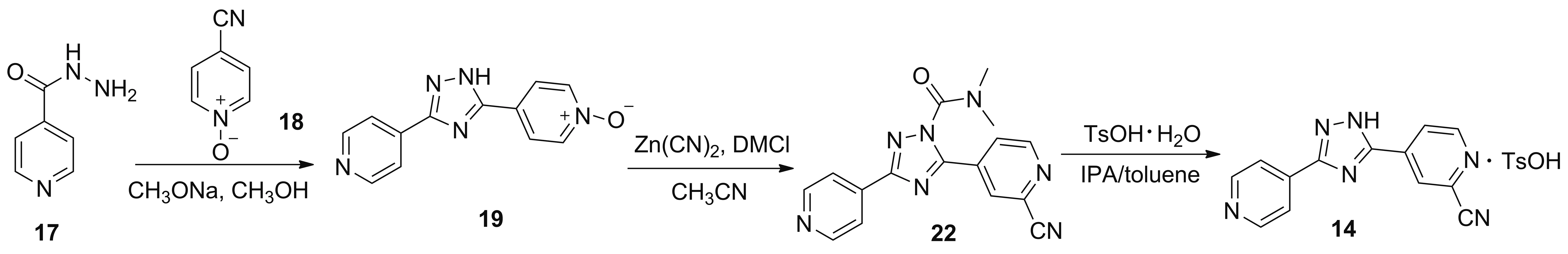
Scheme 14
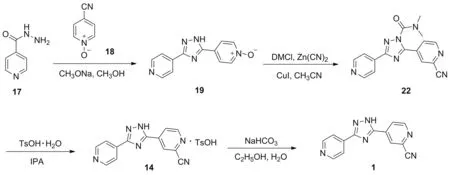
Scheme 15
Scheme 16
2.2 以4-氰基吡啶-N-氧化物(18)为原料
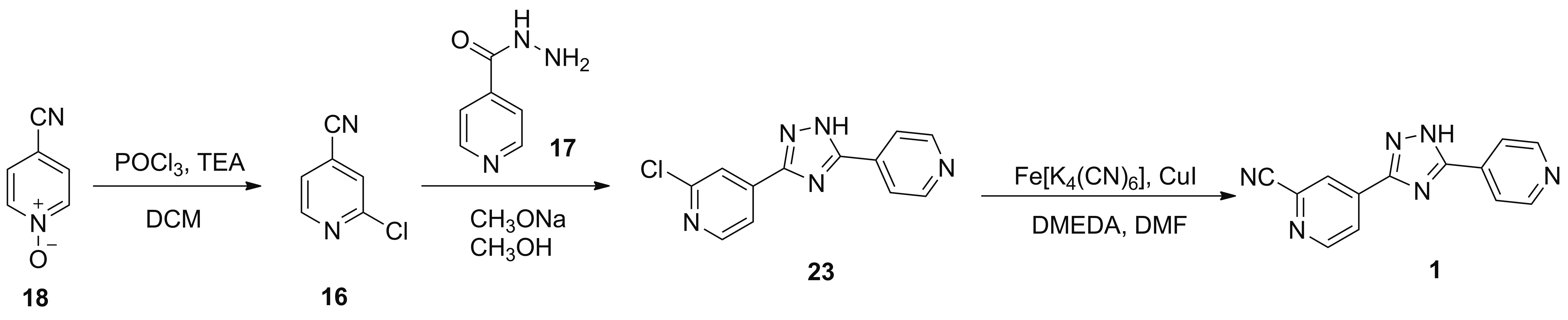
2015年,孙滨等[22]报道了以18为起始原料合成1的工艺路线(Scheme 16)。于DCM中,原料18在POCl3和TEA的条件下发生氯化反应得到16,收率为84.9%; 16与17在甲醇钠/甲醇体系中发生关环反应制得5-(2-氯-4-吡啶基)-3-(4-吡啶基)-1,2,4-三唑(23),收率为90.0%; 23在亚铁氰化钾(1.1 eq.)、碘化亚铜(0.1 eq.)、 DMAC(0.3 eq.)的作用下于DMF中发生氰化反应制得1,收率为81.5%。该路线步骤少,收率高,路线总收率在62.3%以上;使用亚铁氰化钾作为绿色氰源,避免了剧毒氰化物的使用;但原料18价格昂贵,如能开发18的简易合成方法,该路线亦具有工业化前景。
2.3 以异烟酸甲酯-N-氧化物(3)为原料
2016年,张凯等[23]报道了以3为起始原料合成1的工艺路线(Scheme 17)。原料3在甲醇中与水合肼发生肼解反应制得异烟肼-N-氧化物(24),收率为78.4%; 24与6在甲醇钠/甲醇体系中发生关环反应得到19,收率为89.4%; 19与对甲苯磺酸于IPA中反应得到4-(3-(4-吡啶基)-1H-1,2,4-三唑-5-基)吡啶1-氧化物对甲苯磺酸盐(25),收率为87.6%;25在TMSCN和DMCl的作用下发生氰化反应得到目标化合物1,收率为90.4%。该路线总收率较高(55.5%),且分离纯化使用打浆和重结晶的方法,操作简单,但使用对甲苯磺酸精制会产生基因毒性杂质对甲苯磺酸酯;异烟酸甲酯-N-氧化物价格昂贵,工艺成本较高,不适合工业化生产。
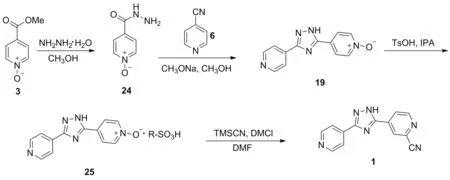
Scheme 17
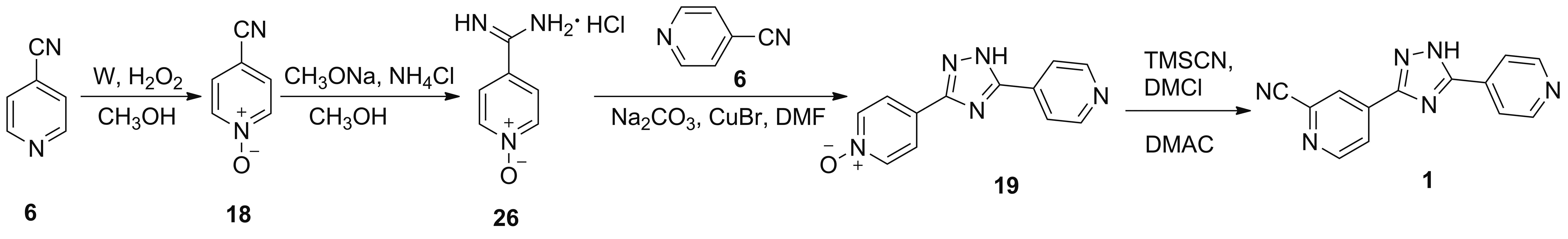
Scheme 18
2.4 以4-氰基吡啶(6)为原料
2017年,乔永辉等[24]报道了以6为起始原料合成1的工艺路线(Scheme 18)。先使钨粉(1% eq.)与双氧水反应制得氧化钨;原料6在氧化钨和双氧水的作用下发生氧化反应制得18;于甲醇中,18在甲醇钠、氯化铵作用下发生脒化和成盐反应制得4-氨基甲酰亚氨基吡啶-N-氧化物盐酸盐(26); 26与6在溴化亚铜和碳酸钠的作用下发生关环反应得到19;于DMAC中,19在TMSCN和DMCl作用下发生氰化反应得到终产物1。该路线操作简单,原料廉价易得,成本低,但第三步反应无收率报道;相比于Scheme 12,若氰化反应能采用亚铁氰化钾等绿色氰源替代TMSCN,则具有较好的工业化前景。
该策略主要为先拼接两个吡啶环(Ⅱ),然后在吡啶2-位引入氰基(Ⅰ),最后关环构建三唑环(Ⅲ)得到终产物1。起始原料主要有:4-氰基吡啶-N-氧化物(18);异烟酸甲酯(12)。
3.1 以4-氰基吡啶-N-氧化物(18)为原料
2013年,富士药品株式会社报道了以18为起始原料合成1的工艺路线(Scheme 19)[25-26]。18与17在甲醇钠/甲醇体系中发生缩合反应得到4-(2-(亚氨基-(4-吡啶基)甲基)肼羰基)吡啶-N-氧化物(27),收率为89.6%;于DMF中,27在氰化钠和DMCl的作用下发生氰化反应得到11,收率为89.7%; 11于2-丁醇/水体系中与磷酸发生关环反应得到产物1,收率为91.5%。该路线的第二步反应采用了剧毒的氰化钠为氰源,两个中间体27和11极性较大,脂溶性差,分离纯化困难;关环反应采用磷酸为缩合剂,需在高温下长时间反应,收率较低;氰基在磷酸和试剂中微量水分的共同作用发生副反应,产生难以除去的杂质C和杂质D(Chart 4),难以达到药品质量的要求。因此,该工艺缺乏工业生产的竞争力。
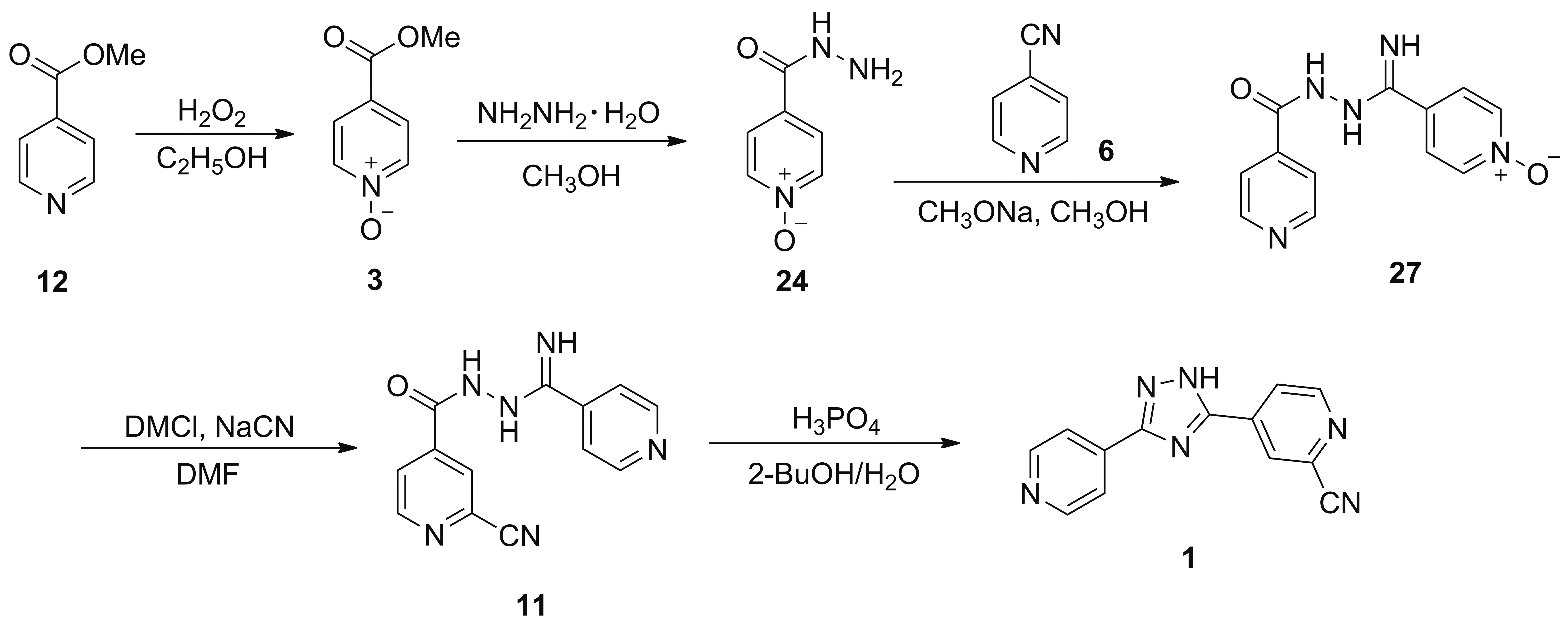
Scheme 20
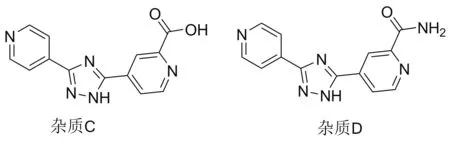
Chart 4
3.2 以异烟酸甲酯(12)为原料
2014年,张涛等[27]在专利中报道了以12为起始原料合成1的工艺路线(Scheme 20),还申请了中间体27的制备方法的专利[28]。原料12在乙醇中与双氧水发生氧化反应得到3,收率为95.2%; 3在甲醇中与水合肼发生肼解反应得到24,收率为96.5%; 24与6在甲醇钠/甲醇体系中发生缩合反应得到产物27,收率为96.8%;于DMF中,27在氰化钠和DMCl作用下发生氰化反应得到11,收率为60.4%; 11于2-丁醇/水体系中与磷酸发生关环反应得到产物1,收率为84.7%。该路线后两步与Scheme 19方法相同,存在一样的弊端,不适合工业生产。
对比以上20条工艺路线,可以发现,制备1的方法有很多,但适合工业化生产的路线并不多见。很多路线都有其各自的亮点,但往往又存在瓶颈和局限。例如,吡啶2-位引入氰基的方法,大多使用剧毒的氰化物作为氰源[29-30],安全隐患和环境污染较大;用酰胺脱水的方法虽然会减少污染,但收率过低。使用三氯氧磷、碘、28%氨水体系引入氰基的方法较新颖,但收率仍有待提高。使用毒性极低的亚铁氰化钾(黄血盐)作为氰源引入氰基的方法,极具吸引力[31]。综合比较之下,孙松等[19]以16为起始原料的工艺路线(Scheme 12)步骤少,收率高,三步反应总收率高达66.8%,且原料廉价易得,工艺成本低;使用亚铁氰化钾作为绿色氰源,避免使用剧毒氰化物,最适合工业化生产。乔永辉等[24]以6为起始原料的合成路线(Scheme 18)操作简单,原料廉价易得,若第四步氰化反应能采用亚铁氰化钾等绿色氰源替代TMSCN,也具有较好的工业化前景。
托匹司他是对痛风有显著疗效的XOR抑制剂,然而该药物在我国至今还未上市。要实现托匹司他在国内的注册申报,其原料药合成工艺研究是当务之急。目前,托匹司他的合成工艺研究还有很大空间。在今后的研究中,可考虑用黄血盐作为绿色氰源,避免剧毒氰化物的使用,降低安全隐患和减小环境污染压力;托匹司他的产品精制,可使用氢溴酸代替对甲苯磺酸,避免基因毒性杂质的产生。此外,如果能实现1,2,4-三唑环与两个吡啶环的高效偶联,托匹司他的合成将会事半功倍。相信随着合成方法学和绿色化学的不断进步,更多简洁高效、绿色环保的先进合成工艺路线会不断涌现。
猜你喜欢
昆明医科大学学报(2021年2期)2021-03-29 07:42:14
理化检验-化学分册(2020年5期)2020-06-15 11:36:08
四川警察学院学报(2019年6期)2019-12-28 07:20:06
当代医药论丛(2017年22期)2017-04-12 06:29:46
文化产业(2016年6期)2016-10-19 19:13:47
国外医药(抗生素分册)(2016年2期)2016-07-12 14:25:05
中国卫生标准管理(2015年4期)2016-01-14 05:16:48
分析测试学报(2015年8期)2016-01-13 06:19:28
淮海医药(2015年2期)2016-01-12 04:33:22
西安建筑科技大学学报(自然科学版)(2014年2期)2014-11-12 13:04:50
