
老年男性排尿障碍9年,发作性意识障碍7年
——神经元核内包涵体病
2020-05-09 08:42:58汪露露王环朱敏洪道俊
中国神经精神疾病杂志 2020年3期
汪露露 王环 朱敏 洪道俊○☆
1 临床资料
患者男,61岁,主因“排尿障碍9年,发作性意识障碍7年”于2018年10月 5日就诊于我院。
患者52岁时出现排尿不畅感,大便干结,曾到医院检查排外了前列腺病变。53岁时发现皮肤干燥,下肢远端前部皮肤出现条纹状色素沉着,伴出汗减少,期间出现发作性发热,常在感冒咽部不适后发生,体温多为37.5~38℃,最高可达39℃,同时出现头痛、全身乏力、淡漠、行走不稳。54岁时因为严重尿潴留而行永久性膀胱造口术,术后当年某日清晨起床时家人发现其神志不清,但当时生命体征平稳,周边无呕吐物,无发热抽搐,送至医院后排除低血糖,给予支持对症治疗,患者数日内逐步清醒。55岁时在一次发热(38℃)后出现意识模糊,伴恶心呕吐及言语和认知障碍,治疗后患者遗留记忆力减退,以近期记忆力为甚,远期记忆力尚可。随后的5年间,共发生类似的发作性意识障碍4次,有时伴发热,有时不伴发热,但均未见肢体抽搐,经过数天的对症治疗后均能清醒。此次再次因出现意识障碍被家人送来我院治疗。近两年来患者记忆力下降、反应迟钝,且逐渐出现偏执、躁狂、胡言乱语、行动迟缓等症状。发病以来饮食尚可,体重无明显变化。
既往史:患者患糖尿病20年,未正规服用降糖药,但平素血糖无显著波动。无特殊药物或毒物接触史。父母身体健康,兄弟姐妹及其他亲属无特殊疾病,家系内无类似疾病患者,否认其他遗传病。
体格检查:正常体型,体温:36.9℃,心率 70次/min,呼吸:15 次/min,血压 105 mmHg/70 mmHg,心律齐,叩诊心界不大,各瓣膜区听诊未闻及杂音,双小腿前部皮肤菲薄,可见紫红色条纹状色素沉着,其他内科查体未见明显异常。嗜睡状态,双侧瞳孔等大等圆,直径约1 mm,对光反射迟钝,双眼球居中位,无明显凝视麻痹,未见眼震。四肢肌肉和躯干肌无明显萎缩,四肢肌力5级。四肢痛刺激回避明显,其他感觉检查不配合。腹壁反射未引出,四肢腱反射未引出。双侧Babinski's征阳性。脑膜刺激征阴性。
辅助检查:血液和大便常规正常,尿常规示白细胞2+,蛋白1+。空腹血糖8.8 mmol/L,肝肾功能、电解质、肌酶谱、血脂,同型半胱氨酸均正常。甲状腺功能正常,自身免疫风湿相关抗体阴性,肿瘤标记物阴性。脑脊液:压力180 cmH2O,白细胞总数 1×106/L(正常值:0~8×106/L),潘氏实验阴性,脑脊液蛋白 516 mg/L(正常值:150~450mg/L),脊液糖 3.99 mmol/L(正常值:2.8~4.5 mmol/L),脑脊液氯125.7 mmol/L(正常值:120~132 mmol/L),脑脑脊液革兰染色、改良抗酸染色、墨汁染色、脱落细胞学均阴性;自身免疫性脑炎抗体全套阴性。心电图未见明显异常。心脏、肝、胆、胰、脾、双肾彩超未见异常。胸腹部CT示:① 双肺多发性陈旧性病灶;两肺多发小结节。② 两侧胸腔少量积液。③ 胆囊高张,胆汁淤积可能,余腹部未见明显异常。3 h视频脑电图示:左侧额叶个别尖波,背景慢活动。55岁发作时头颅MRI示胼胝体压部DWI和Flair高信号 (图1A);57岁发作时头颅MRI示双侧额顶颞叶皮髓交界区及胼胝体散在DWI高信号(图1B);58岁发作时头颅MRI示双侧额顶叶皮髓交界区线状,及胼胝体DWI高信号,伴左侧颞顶叶皮髓水肿(图1C),半年后再次发作显示皮髓交界区病灶仍存在,但左侧颞顶水肿病灶部分消退,伴皮髓区DWI高信号(图1D);60岁发作时头颅MRI显示额顶叶皮髓交界区病灶仍然存在,伴脑白质病变改变(图1E);此次入院(61岁)头颅MRI显示额顶叶皮髓交界区病灶往后部延伸,脑白质病变较前加重(图1F)。
病理检查:经知情同意,患者行左下肢外踝上10 cm处皮肤活检。HE染色光镜显示皮肤构筑结构正常,在汗腺导管的上皮细胞核内可见嗜酸性包涵体(图2A),在成纤维细胞和脂肪细胞核内也可见嗜酸性包涵体(图2B)。p62蛋白免疫组化染色显示汗腺导管上皮细胞(图2C)和脂肪细胞核内的嗜酸性包涵体阳性表达。皮肤电镜检查显示汗腺导管上皮细胞(图3A)和成纤维细胞(图3B)核内出现无膜包裹的8~10 nm细丝样物质沉积。
基因检查:运用重复引物PCR(RP-PCR)法检测显示NOTCH2NLC基因的 5'不翻译区(5'UTR)的 GGC重复扩增呈递减的锯齿样延伸,初步估算超过100次(图4A),而对照者没有锯齿样改变 (图4B)。进一步荧光扩增长度PCR (AL-PCR)法检测显示NOTCH2NLC基因5'UTR区GGC重复扩增为121次 (图4C),正常对照为16次 (图4D)。根据临床、影像、病理和基因检测结果,诊断神经元核内包涵体病 (neuronal intranuclear inclusion disease,NIID)。

图1 头颅MRI A:患者55岁发作时头颅MRI示胼胝体压部DWI和Flair高信号;B:57岁MRI示双侧额顶颞叶皮髓交界区及胼胝体散在DWI高信号;C:58岁MRI示双侧额顶叶皮髓交界区线状,及胼胝体DWI高信号,伴左侧颞顶叶皮髓水肿;D:半年后再次发作显示皮髓交界区病灶仍存在,左侧颞顶水肿病灶部分消退,伴皮髓区DWI高信号;E:60岁MRI显示额顶叶皮髓交界区病灶仍然存在,伴脑白质病变改变;F:61岁MRI显示额顶叶皮髓交界区病灶往后部延伸,脑白质病变较前加重

图2 皮肤活检HE染色 A:光镜显示皮肤汗腺导管上皮细胞核内可见嗜酸性包涵体;B:在脂肪细胞核内也可见嗜酸性包涵体;C:p62蛋白免疫组化染色显示汗腺导管上皮细胞和脂肪细胞;D:核内的嗜酸性包涵体阳性表达
治疗:患者此次入院后给予抗生素(尿路感染),补液营养支持治疗,患者在入院后3 d基本能够下床行走生活自理。6 d后出院时进行认知功能评定显示MMSE评分量表13分,MoCA评分量表16分,FAB评分量表6分,提示重度认知障碍,额叶损害较重。患者继续服用盐酸多奈哌齐和多种维生素,加强尿路管理。2020年3月随访,患者未再发作意识障碍。

图3 皮肤电镜检查 汗腺导管上皮细胞(A,箭头)和成纤维细胞(B,箭头)核内出现无膜包裹的细丝样物质沉积
2 讨论
神经元核内包涵体病又称神经元核内嗜酸性包涵体病,是一种罕见的神经退行性疾病[1-2]。1968年LINDENBERG[3]在1例28岁神经变性病患者的神经细胞和内脏细胞内发现了核内包涵体。1984年HALTIA[4]进一步发现其病理特征是神经元大量丢失,神经细胞以及全身多个其他器官中存在嗜酸性核内包涵体,免疫组织化学显示包涵体泛素阳性,电镜显示核内出现无膜包裹的细丝物质沉积,故命名为NIID。
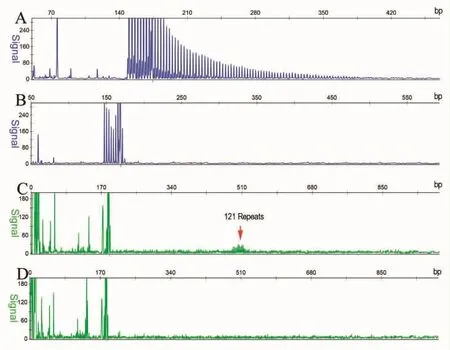
图4 重复引物PCR NOTCH2NLC基因的5'不翻译区(5'UTR)的GGC重复扩增呈递减的锯齿样延伸,初步估算超过100次(A),对照者没有锯齿样改变 (B)。 荧光扩增长度PCR显示NOTCH2NLC基因5'UTR区GGC重复扩增为121次(C),正常对照为16次(D)
NIID临床表型具有高度的异质性,根据发病年龄可以分为婴幼儿型NIID、青少年型NIID、成年型 NIID,我国以及东亚地区主要以成年NIID为主[1,5-8]。文献显示成年型NIID发病年龄有两个高峰期,以肢体无力为主要表现的在30岁左右发病,以认知障碍为主要表现的在55岁左右发病。我们的研究显示多数的成年型NIID表现为认知障碍为主,就诊时间在60岁左右,患者就诊的主诉多为发作性脑病、排尿障碍、肢体震颤、偏头痛发作和视力障碍等,就诊后仔细排查才发现患者存在认知障碍[1]。此例患者表现为发作性脑病,具有很强的诊断提示意义,其发生率从10%~65%不等,患者多在一些应激事件后出现,特别是长期导尿后反复尿路感染诱发,可以表现为意识障碍、意识淡漠、精神症状、头痛呕吐、虚弱等,临床上仅需要给予抗生素和对症治疗即可,患者很快能恢复到基线状态[1-2]。此外,多数患者表现为不明原因的小便排解困难,部分患者作为首发症状早于其他症状多年出现,并最终进展为留置导尿或膀胱造口,临床上对于突出的排尿障碍要警惕NIID的可能[1,3]。因此,我国成年型NIID患者临床特征为大脑功能障碍、自主神经神经病变与周围神经病变的临床组合[1]。
成年型NIID患者的头颅磁共振成像(MRI)显示额顶颞叶皮髓交界区DWI线样的高信号,随着病情发展,从大脑前部向后部逐步延伸,形成“鸡冠花样”的DWI高信号。这种选择性累及弓形纤维,不易消退的DWI高信号病灶具有一定的特征性,也是目前国内诊断NIID被广泛认识的主要原因[1-2]。然而,成年型NIID家系内研究显示仅37.5%患者出现上述DWI改变[9],我们的研究也显示部分患者并不出现皮髓交界区DWI高信号,提示如果过份依赖该影像特征可能导致NIID的漏诊。此例患者首先出现胼胝体压部的DWI高信号,病情发展后出现皮髓交界区DWI高信号,类似的现象在其他患者中也可观察到,提示胼胝体联络纤维和皮层下弓形纤维具有类似的易受累性[1]。此外,对称性的均匀弥漫的脑白质病变也是成年型NIID的MRI改变之一,已有研究显示,在不明原因的成年型脑白质病变中,NIID是继CADASIL之后第二常见的原因[10]。
NIID其疾病名称本身就是一个病理学意义上的诊断,因此病理上发现核内包涵体对NIID的诊断非常重要[1-2]。在该病研究的早期阶段,主要通过尸检获得病理确诊,研究发现核内包涵体不仅出现在神经元内,也可以出现在胶质细胞、结肠组织神经节细胞、心肌细胞、骨骼肌细胞、肾小管上皮细胞、周围神经雪旺细胞等全身多种组织[2]。这些包涵体内出现多种蛋白表达,特别是出现泛素和p62蛋白表达时,具有一定的诊断价值。电镜下这些包涵体为一堆直径在8~10 nm的细丝沉积物质。2011年日本SONE[11]发现NIID患者皮肤的汗腺导管上皮细胞、成纤维细胞、脂肪细胞核内存在嗜酸性性包涵体,具有脑病理等价值的诊断意义。此后,NIID的诊断得到了较快的发展,特别是在国内逐步有较多患者通过皮肤活检得到确诊。我们的研究显示:只要具有典型的临床表现,在皮肤活检标本的光镜下发现嗜酸性包涵体,且p62阳性标记,即可以确诊NIID,而不需要进一步电镜检查的支持[1]。
1984 年HALTIA报告的NIID病例就是一个双胞胎患者,随后国内外很多NIID患者出现家系患者,提示NIID可能和基因变异有关。然而NIID患者既有常染色体隐性遗传患者,也有常染色体显性遗传患者,更有很多的散发患者,所以长期以来NIID的基因定位没有成功。2019年日本的两个团队[12-13]、湘雅医院唐北沙团队[9],以及我们团队[14]几乎在同一时间通过三代测序发现,人类特异性NOTCH2NLC基因5'区GGC异常重复扩增,是成年型NIID的致病基因,我们团队进一步证实青少年型NIID也是该基因变异导致。目前研究显示NOTCH2NLC基因5'区GGC异常重复扩增次数超过66次就可能致病[9],该例患者经过RP-PCR和AL-PCR证实其GGC重复次数达121次,因此,我们报告的这例患者具有典型的临床表型,也具有明确的病理改变和基因变异。
成年型NIID是一种进行发展的神经系统变性病,部分患者伴随间断的发作加重,目前尚无有效的特异性治疗。对于发作性脑病的治疗,目前不同学者有不同的认识,有的主张在发作期使用激素治疗,有的认为不需要特殊治疗。我们的观察提示急性发作的脑病只需对症支持处理即可,患者几乎在数日内即能恢复到基线水平[1-2]。需要强调的是尿路管理,我们的研究发现部分长期留置尿管或者膀胱造瘘患者反复发生尿路感染,在此基础上诱发脑病发作。
3 点评
作为一个神经遗传病学者,最让人兴奋的莫过于疾病新基因的克隆和临床表型的扩展。神经遗传病学发展至今,尽管中国学者首先定位克隆的基因仍然相对较少,但近年来我国学者正在大步赶上。本专栏前一篇家族性发作性疼痛综合征3型(FEPS3)的致病基因SCN11A就是由华中科大协和医院王涛教授团队首先克隆,而本文NIID的致病基因最早文献见刊的是湘雅医院唐北沙教授团队。此外,唐北沙教授团队和我们的团队进一步发现NOTCH2NLC基因5'区GGC异常重复扩增是部分特发性震颤[15]、阿尔茨海默病[16]、帕金 森病[9]、多系统 萎缩[17]、成年脑白质病[10]、运动神经元病[9]等多种神经系统变性病的致病原因,随着基因型和临床型的逐步明确,学界目前建议将NIID疾病名变更NOTCH2NLC重复扩增相关性疾病(NOTCH2NLC-related repeat expansion disorder)[18]。
猜你喜欢
中国临床医学影像杂志(2022年6期)2022-07-26 07:17:14
法人(2021年12期)2021-05-09 17:24:24
保健与生活(2019年11期)2019-07-31 01:53:16
中国医学影像技术(2019年3期)2019-03-25 03:45:52
武警医学(2018年10期)2018-11-06 07:04:36
大连医科大学学报(2018年6期)2018-04-11 04:34:48
中国CT和MRI杂志(2016年11期)2017-01-18 10:57:05
安徽农业科学(2015年10期)2015-04-24 08:19:26
新农村(2014年12期)2015-04-08 02:32:48
河南医学研究(2014年3期)2014-02-27 14:51:51
