
UPLC-Q-Exactive测定茶叶中的氟虫腈及其代谢物
2020-05-09 08:12:10李宇翔李燕平李少光
福建医科大学学报 2020年1期
李宇翔, 李燕平, 李少光, 杨 艳
中国是世界上第一产茶叶大国,茶叶产品畅销国内外。但是近年来茶叶农药残留问题日益严重,同时国际市场尤其是欧盟和日本这两大市场却启用了更严格的农药残留限量标准,使农药残留成为茶叶出口的主要限制因素。虽然氟虫腈在茶树上未被登记使用过,但却在出口茶叶中多次检出[1]。2017年荷兰毒鸡蛋事件发生后,氟虫腈成为欧美国家重点关注的农药残留之一,日本市场甚至对我国的乌龙茶实施氟虫腈重点检查[1]。
氟虫腈是一类苯基吡唑类广谱杀虫剂,由于其具有杀虫类别广,杀虫效果好,时效长,不影响作物生长等优势被广泛用作多种作物的杀虫剂[2]。氟虫腈稳定性较差,在使用过程中会产生氟甲腈、氟虫腈亚砜、氟虫腈砜等。氟虫腈及其代谢物的大剂量的摄入会造成人体肝功能、甲状腺及肾脏损伤[3]。研究表明,作为代谢物之一的氟虫腈砜对淡水生物的毒性比氟虫腈高6倍以上[4]。因此,测定氟虫腈的同时也需要测定其代谢产物。
我国也逐渐重视氟虫腈的测定。目前文献报道测定茶叶中氟虫腈的方法主要有气相色谱法、气相色谱-质谱联用法、高效液相色谱法、高效液相色谱-串联质谱法等[4-10]。文献还未发现UPLC-Q-Exactive法测定茶叶中的氟虫腈及其代谢物。因此,本研究旨在建立运用较为成熟的QuEChERS(Quick,Easy,Cheap,Effective,Rugged,Safe)农药残留快速检测方法[11-12],与UPLC-Q-Exactive仪器联用,通过一级全扫(full mass spectrometry/target-select iron monitoring,Full MS/t-SIM)定量,二级平行监测(parallet-reaction-monitoting,PRM)进行定性分析,快速、准备、灵敏地检测和筛查出茶叶中氟虫腈及其代谢物残留。
1 材料与方法
1.1试剂与仪器
1.1.1试剂 氟虫腈标准品(批号:7783741)、氟甲腈标准品(批号:4427363)、氟虫腈砜标准品(批号:160021)、氟虫腈亚砜标准品(批号:4092033)均购自美国迪马公司;氯化钠(批号:20170413)、无水硫酸钠(批号:20180615)均购自国药集团化学试剂有限公司;N-丙基乙二胺(PSA)(批号:030982-TE)、C18粉(批号:030982-TE)均购自美国Thermo公司;乙腈、甲醇均购自德国默克公司。
1.1.2仪器 液相色谱质谱联用仪(Thermo UltiMate 3000 UPLC-Q Exactive,美国Thermo公司);离心机(ALLEGRA X-30,美国贝克曼库尔特公司);纯水机(MILLIPORE Milli-Q8,美国Milli-pore公司);数控超声波清洗器(KQ-250DE型,昆山市超声仪器有限公司)。
1.2方法
1.2.1样品处理 通过优化文献方法[13],将商品化的茶叶样品搅碎至粉末状,再称取茶叶样品2 g于50 mL离心管中,加20 mL乙腈,涡旋混匀30 s,超声提取15 min,加入2 g氯化钠和6 g无水硫酸钠,涡旋2 min,9 000 r/min离心10 min,取上清液1 mL,加入50 mg PSA粉末、50 mg C18粉末和250 mg无水硫酸钠,涡旋30 s,10 000 r/min离心5 min,取上清液0.5 mL加水0.5 mL,涡旋30 s,过0.25 μm有机滤膜,待测定。
1.2.2标准溶液的配制
1.2.2.1氟虫腈及其代谢物标准储备液的配制 分别精密称取氟虫腈、氟甲腈、氟虫腈砜、氟虫腈亚砜标准品0.01 g,用乙腈溶解并定容于100 mL量瓶,终浓度为100 mg/L。氟虫腈及其代谢物混合标准使用液的配制:准确称取0.1 mL标准储备液(100 mg/L),用乙腈溶解并定容于100 mL量瓶,终浓度为100 μg/L。
1.2.2.2氟虫腈及其代谢物混合标准曲线的配制 取适量混合标准工作液,用空白基质溶液配制成浓度为0.1,0.2,0.5,1,2 μg/L的标准工作曲线,临用现配。
1.2.3仪器条件
1.2.3.1色谱条件 色谱柱:Thermo Syncronis C18柱( 100 mm×2.1 mm,1.7 μm );流速0.3 mL/min;柱温40 ℃;进样量10 μL;流动相 A为甲醇,B为纯水溶液,梯度洗脱程序:0 ~0.5 min,90%A;0.5~2.5 min,90% ~ 5%A;2.5~4 min,5%A;4~4.1 min,5%~90%A;4.1~6 min,90%A。
1.2.3.2质谱条件 扫描模式1:Full MS/t-SIM(150~2 000 m/z);分辨率:70 000;采集极性:Negative;AGC target:1e6;Maximun IT:50 ms;电喷雾离子源(HESI-Ⅱ);毛细管温度320 ℃;喷雾电压:3.20 kV;鞘气流速:40 arb;辅气流速:15 arb。扫描模式2:PRM(200~800 m/z);分辨率:17 500;采集极性:Negative;AGC target:2e5;Maximun IT:100 ms。
2 结 果
2.1氟虫腈及其代谢物的定性鉴定 通过Full MS/t-SIM和PRM对氟虫腈标准品进行定性考察,获得其实际精确母离子和子离子质核比信息、色谱峰的保留时间,快速初筛出空白基质样品[14]。两种模式下基质空白图谱见图1,2,未检出任何峰型信息。氟虫腈及其他3种化合物的相关文献信息及质谱测定信息见表1。测得实际精确分子量与理论精确分子量的偏值均<5×10-6,可以确认为目标化合物。

表1 氟虫腈及其代谢物高分辨质谱鉴定参数表
△:质量偏差P等于理论值T与实际值的差值与理论值的比值.
2.2线性、检出限及定量限 按照1.2项下方法做基质标准曲线,线性范围为0.1~2 μg/L,其中Full MS Scan Type(线性ya)的相关系数为0.996 0~0.997 6,PRM Scan Type(线性yb)的相关系数为0.995 9~0.999 8。计算当信噪比为3倍(S/N≥3)时的溶液浓度为检出限,信噪比为10倍(S/N≥10)时的溶液浓度为定量限。两种不同检测模式的检出限和定量限见表2。两种扫描模式的标准加标色谱及质谱图见图3,4。
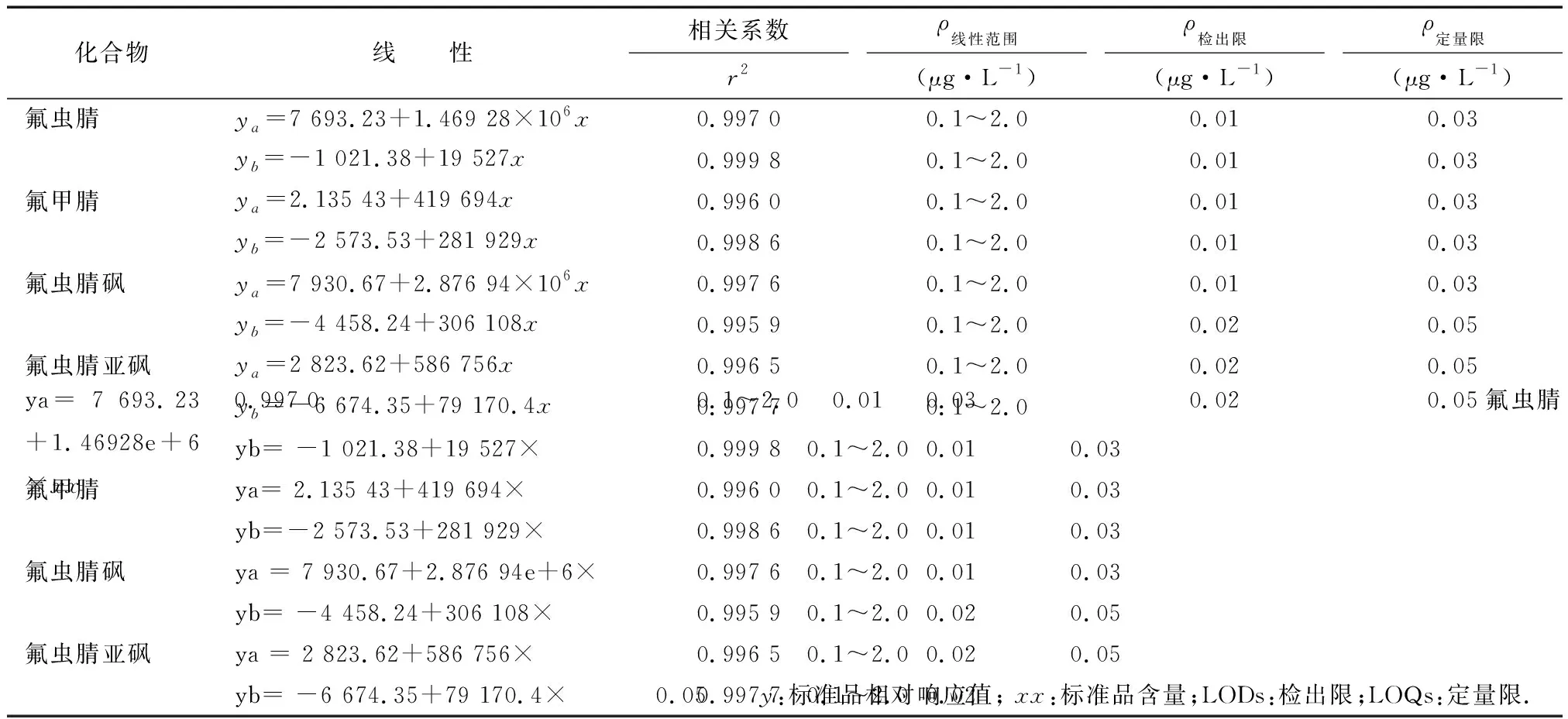
表2 氟虫腈及其代谢物线性关系、范围、检出限以及定量限
y:标准品相对响应值;x:标准品含量;LODs:检出限;LOQs:定量限.
2.3回收率、精密度 通过使用前期筛查的空白基质样品进行加标回收率试验,每种化合物分别添加3种不同浓度的标准物质(0.5,1.0,2.0 μg/kg),每个平行测定6次(n=6),按2.1项下处理方法进行处理,并进质谱分别进行Full MS/t-SIM和PRM模式的定性定量分析,结果见表3。Full Mass扫描模式下测得氟虫腈及其代谢物的平均回收率为90.2%~106.7%,RSD值为2.035%~5.438%;PRM模式下的平均回收率为92.0%~103.2%,RSD值为1.075%~5.930%。
2.4实际样品测定 按食品抽样方法,随机抽取全省190份茶叶样品,按1.2.1~1.2.3项下处理方法对样品进行测定。用氟虫腈及其代谢物的标准品进行定性定量。结果表明,190份样品检出43份(按本方法的检出限计算,包含超出国家限量指标),一级母离子精确质量偏差<5×10-6,阳性样品Full MS质谱图见图5,质谱图见图6。
表3 氟虫腈及其代谢化合物的回收率、精密度
Tab 3 The average recovery, precision of fipronil and its metabolites in tea

化合物加标浓度(μg·kg-1)平均回收率/%Full MS PRM相对偏差RSD/%Full MSPRM氟虫腈0.592.995.43.7493.4581.090.292.02.0351.6212.094.195.63.0675.930氟甲腈0.596.992.33.1102.6531.091.795.82.7621.0752.090.793.82.6081.846氟虫腈砜0.596.4101.94.1701.5741.0103.097.05.4382.4182.0105.9101.03.8323.748氟虫腈亚砜0.597.993.42.8193.6031.0101.4103.03.7425.0182.0106.7103.24.9032.809
n=6. Full MS:一级全扫;PRM:二级平行监测.
3 讨 论
由于食品基质的复杂性及涉及多组分、同类型化合物结构的相似性,普通低分辨率串联质谱仪无法准确分离定性,因而常出现假阳性。传统的检测方法已无法满足日常的检测要求。高分辨质谱仪在一定程度上弥补了这个缺陷,被广泛地应用于农药的快速筛查[15]。Q Exactive高分辨质谱仪通过四极杆与Obitrap串联,提高了定性能力,同时其定量能力达到了三重四极杆的定量效果[16]。本方法基于四极杆-静电场/轨道阱质谱的高分辨能力,可有效地排除样品基质效应的影响,与传统三重四极杆质谱仪相比,其分辨率高,对化合物的质核比能够精确至小数点后5位,具有排他性,因而可以进行准确的一级定量。同时本方法也对样品进行PRM模式的测定,通过对阳性样品精确的二级子离子扫描(子离子m/z精确至小数点后5位),进一步对目标化合物进行确证,从而消除假阳性的可能性。两种扫描模式的联合运用,保证了方法的定量准确和定性可靠。结果表明,本方法分析时间短,灵敏、准确,检出限均能达到0.02 μg/L,可以满足各种茶叶中氟虫腈及其3种代谢物的快速测定要求,有一定的推广价值。但是由于本研究筛查组分少,未考虑流动相及色谱柱的选择,并且氟虫腈及其代谢物在4 min同时出峰,考虑后期加入更多的组分,在之后的试验应逐步优化色谱条件,提高仪器的效率及分辨能力。
猜你喜欢
化工设计通讯(2022年10期)2022-12-31 20:42:50
现代临床医学(2022年4期)2022-09-29 07:36:10
波谱学杂志(2022年2期)2022-06-14 09:52:02
分析仪器(2019年3期)2019-06-18 08:38:58
农药科学与管理(2019年2期)2019-01-05 14:01:42
环球时报(2017-08-03)2017-08-03 11:55:02
化工科技(2016年6期)2016-06-06 01:54:24
分析测试学报(2015年7期)2016-01-13 06:19:16
质谱学报(2015年5期)2015-03-01 03:18:37
现代检验医学杂志(2015年1期)2015-02-06 01:59:14
