
化学计量学方法在脂质组学数据解析中的应用
2020-05-08 13:40覃佐剑
分析测试学报 2020年3期
覃佐剑,谢 亚,魏 芳,陈 洪
(中国农业科学院油料作物研究所,农业农村部油料加工重点实验室(武汉),脂质化学与营养 重点实验室(湖北),湖北 武汉 430062)
脂质组学研究中,一方面,由于样本中不同的脂质种类浓度通常呈数量级级别差异且杂质干扰严重,研究者需根据样本特点,采取有效的脂质样本前处理方法(如液液萃取、固相萃取等),将待测脂质从生物样本提取出来[7-8],并针对不同类型样本,选择合适的质谱检测技术(如直接进样技术、色谱-质谱联用技术),优化色谱洗脱条件、质谱离子化方式和数据采集模式(如产物离子扫描、中性丢失扫描、多反应监测等),以获得高质量的原始数据,为脂质组学数据解析奠定基础。另一方面,采用合适的化学计量学方法和软件包来解析脂质信号,是获得准确分析结果的重要保证。在此基础上,结合“脂质代谢物和代谢途径研究策略”(Lipid metabolites and pathways strategy,LipidMAPS)[9]、格拉茨理工大学脂质数据库(LipidHome)[10]、人类代谢物数据库(Human metabolome database,HMDB)[11]、加州大学戴维斯分校脂质计算数据库(LipidBlast)[12]、京东大学基因及基因组百科全书(Kyoto encyclopedia of genes and genomes,KEGG)[13]、美国斯坦福研究院脂质实验数据库(MetCyc)[14]等多种脂质结构和代谢通路数据库,可实现脂质的定性定量分析和代谢通路研究。针对脂质组学复杂数据的处理问题,不同研究团队已经开发出了多种集成化学计量学算法的脂质组学数据分析软件和方法包,如XCMS[15]、MZmine[16]、OpenMS[17]、LipidDataAnalyzer[18]、LOBSTAHS[19]、LipidHunter[20]等。这些算法和工具包的使用,在一定程度上降低了数据解析的难度,提高了脂质组学数据处理的能力。
脂质组学数据的预处理和统计分析是降低数据复杂度、提取特征脂质信号,实现脂质定性定量、挖掘脂质生物标记物和样本生物学意义的关键步骤。研究表明,化学计量学在脂质组学数据的基线校正与背景扣除、信号峰识别、保留时间校正、同位素分布解析等领域得到了广泛应用[15,18,20-24]。针对脂质组学复杂数据的解析问题,研究人员已建立了如小波变换[25]、多项式拟合[26]、非负最小二乘法[27]、去卷积算法[28]、机器学习[29]等大量的化学计量学算法。同时,随着新型多维高通量仪器的使用,脂质数据更为复杂,分析难度进一步增大,新的化学计量学方法和数据处理策略成为研究人员关注的热点。化学计量学在脂质组学数据分析中的应用主要分为背景及基线校正(Baseline correction and background noise deduction)、信号峰识别(Signal peak detection)、保留时间校正(Retention time alignment)、同位素分布解析(Isotope distribution resolving)和统计学分析(Statistical analysis) 5个部分(图1)。本文就近年来化学计量学在脂质组学数据解析中的应用进行了综述。

图1 脂质组学数据分析流程
Fig.1 Workflow of lipidomics data processing
1 色谱基线校正与背景噪声扣除
在脂质组学原始数据采集的过程中,因为固定相的性质差异和流动相化学组分的改变,经常会产生基线漂移和背景噪声,这些不稳定因素会严重干扰数据的后续分析。因此,有效的基线校正和背景扣除对于脂质数据的准确分析,具有十分重要的意义。研究表明,基于化学计量学的基线校正和噪声扣除算法,能够有效地提高数据处理的自动化程度和定性定量的准确性[26,30-36]。
对于色谱背景频率基线呈常数的情况,可以应用空白对照谱图进行简单校正,但通常色谱背景由于程序升温或者流动相梯度等因素的改变,大多呈现非线性变化,因此应用该方法会产生较大偏差。针对非线性变化的复杂背景基线校正,Windig等[30]开发了基于均值扣除的成分检测算法(Component detection algorithm,CODA),该算法先对色谱峰背景进行均值计算,自动扣除低于均值的噪声信号,再通过引入相似系数对处理结果进行验证筛选,实现背景噪声的扣除和基线校正。但该方法准确度不高,难以捕捉高度相关样品的细微差别。改进的CODA算法,先通过标准计算自建模交互技术的伪秩,确定信号的连续性,再依据实际信号与筛选信号的峰宽相似度进行噪声过滤和背景校正。该法提高了对重复样本的处理能力,但对于信号强度接近背景噪声的组分信号,依然不能进行有效处理[37]。另一类是基于多项式拟合的基线校正方法。简单的多项式拟合并不能解决复杂的基线漂移问题,中山大学甘峰团队[26]结合递归法提出了改进的多项式拟合法,该法首先应用多项式拟合基线噪声对未拟合的部分自动生成阈值,然后通过迭代算法重复优化预测模型,最终实现较为准确的基线校正和背景噪声过滤。经比较分析,可对正常信号实现准确识别,在针对包含重叠峰、拖尾峰等复杂的色谱信号时,也有较好的效果。小波变换算法在处理以时间和频率为窗口的色谱信号方面具有较大的优势,被广泛应用于色谱的噪声抑制、色谱信号识别、背景校正等[38]。经典的小波变换算法,过于依赖滤波器和参数的手动选择,并不适用于复杂数据的快速处理。Liu等[39]在经典小波变换的基础上,提出最小均方(Least mean square,LMS)的自适应小波变换。该方法运用算法实现滤波器的自动选择,对多种色谱信号均具有较好的兼容性,有效地解决了因小波变换滤波器选择导致的分析结果不稳定的问题。Bertinetto等[40]提出了基于迭代拟合的连续小波变换(Continuous wavelet transform,CWT)的基线自动识别算法,可自动选择和拟合具有代表性的典型数据点,实现大量图谱的快速处理。与一般的小波变换对比表明,该方法解决了参数设置复杂和耗时长的问题,可实现背景基线的快速准确识别,已用于人类卵巢癌细胞样本中磷脂酰胆碱(Phosphatidylcholine,PC)、磷脂酰乙醇胺(Phosphatidyl ethanolamine,PE)、鞘磷脂(Sphingomyelin,SM)等脂质代谢物的研究分析[41]。
此外,针对高分辨率仪器产生的复杂数据基线校准和背景扣除问题,研究者提出了一些基线校正和背景扣除的新方法。如Liu等[42]为解决背景噪声对待测组分的干扰,开发了基于双边指数自动校正算法(Automatic two-side xponential baseline correction algorithm,ATEB),该算法除可有效地校正基线之外,还能通过多次平滑消除异常值的影响。Filgueira等[36]结合多种一维色谱基线漂移校正方法,提出了LC×LC色谱联用的二维背景校正算法(Orthogonal background correction,OBGC),并将其用于二极管阵列背景信号的解析。
2 信号峰识别
信号峰识别是指从噪声干扰严重的原始数据中提取有效的脂质组分信号。由于生物样本(如血浆、肝脏等)中的脂质种类多、脂质同分异构和同重现象普遍等内部因素及仪器不稳定、信号响应差异大等系统因素,使得原始数据中假峰、重叠峰等普遍存在。因此,复杂样本中脂质组分信号的准确识别已成为数据解析的一大难点。基于化学计量学的信号自动识别算法,在复杂脂质信号识别处理方面已取得了重要进展[43-50]。
基于一维质谱的脂质组学分析方法,能够根据分子极性差异实现不同脂类的快速高通量分析。对于一维的质谱信号解析算法,主要有局部最大值(Local maximum)、递归阈值(Recursive threshold)、信噪比过滤(Signal noise ratio filtering,SNR)、连续小波变换(Continuous wavelet transform,CWT)等[43]。局部最大值法通过检测原始数据中的局部最大值,来识别组分的质谱信号,该法简单直接,已被安装在LIMSA[44]、MZmine[16]、mMass3[45]等多个软件工具中。递归阈值法类似于局部最大值法,前者适合于不需要平滑处理的数据,而后者对在连续扫描下采集的原始数据处理效果更好[16]。但这两种方法对低丰度组分信号识别准确度不高。SNR阈值过滤法是通过预先设定的阈值来对一定信噪比强度的峰进行识别筛选,但在识别较弱的脂质信号时,需调整相关阈值。该法在一定程度上解决了低丰度信号的识别问题,并已被应用于脑脊液样本的研究以发现与神经类疾病相关的脂类生物标记物[46]。与SNR阈值过滤法不同,Lange等[47]提出应用CWT算法来解决质谱信号的解析问题,其先将谱图分段处理,然后在确定的分段尺度下利用CWT来检测峰值并估计峰值参数,实现信号识别。该法适用于噪声信号复杂的样本数据,但需选择合适的分段尺度才能达到理想的识别效果,分段处理的不同会导致结果有较大差异。Du等[48]在CWT算法基础上结合信号峰形参数,开发了2D-CWT信号峰自动识别算法,先将小波变换到小波空间中,再利用小波系数中附加的峰形参数进行优化处理,这样可以大大提高信号的有效信噪比,也可实现低丰度信号的有效检测。对一维的质谱数据进行解析时,基于以上方法,能够对峰形好、丰度高的组分信号进行有效的处理,但对于低信噪比的信号需手动优化参数。而对于重叠严重的复杂脂质信号识别问题,还需借助多维的分析技术和算法来实现准确的解析。
二维色谱-质谱联用脂质分析技术,结合了色谱的分离能力和质谱的高选择性,在复杂脂质的有效分离和高通量分析上具有独特优势,已成为脂质组学研究的主要手段之一。针对二维技术的复杂信号分析,Hastings等[49]基于保留时间(Retention time,RT)和质谱质荷比(m/z)构建了数据对矢量化峰值检测算法,通过色谱和质谱信号的一一对应,来有效避免噪声,实现组分信号的准确识别。Andreev等[50]开发了噪声过滤扣除算法(Matched filtration with experimental noise determination,MEND)来解决LC-MS的信号解析问题。Zhang等[51]开发了基于降维的PeakID算法,用以解决多维信号的处理问题,可实现高维复杂数据的快速分析。Yu等[29]开发了基于机器学习的信号识别策略,该方法先通过对原始数据的自适应分段处理,得到包含特征信号的提取离子谱(Extracted ion chromatograms,EICs),再通过逻辑回归(Logistic regression)、支持向量机(Support vector machine,SVM)、随机森林(Random forest,RF)等多种分类模型对EIC进行筛选,最后借助交叉验证模型处理,即可得到匹配度最好的提取谱图,用于后续的定性定量分析;该方法对二维及以上的复杂色谱数据处理具有独特优势。Collins等[19]基于脂质加合物离子的m/z、RT等多种特性,提出了一种正交识别算法(Orthogonal screening criteria),并将其用于硅藻样本中脂质、氧化脂质、脂质生物标志物的自动分析,已成功地从海洋硅藻样本的21 869特征峰中准确鉴定了1 969种核心脂质。
3 色谱保留时间校正
在脂质组学分析研究领域,脂质分子的色谱保留时间与分子极性、脂肪酸酰基链长度和不饱和程度密切相关。脂质数据的色谱保留时间校正是实现多样本平行分析、脂质组分差异判断的重要依据[52-54]。在实验操作中,因色谱柱老化、分析物互相反应、检测器非线性响应等不可控制因素,色谱图会出现色谱峰漂移现象,干扰脂质组分的分析,这就需要借助保留时间校正对其进行校正。脂质组学研究中,保留时间校正通常有内标校正和保留时间校正两种策略。
而除了袖子里能放一些小物件外,汉服的胸前也是可以放东西的。古人的着装要求一般都会把上衣掖在中裤或下裳里(就像现代规范的衬衫要塞在裤子里一样)。这样,腰带与交领结合处就形成了一个三角形口袋。只要这个东西不大、不滑、不细、不硬就可以放在里边了。由于是交领右衽,所以拿取物品很是容易。我们现在所说的“怀揣”就是指这个地方。
内标校正法即在样本预处理过程中,向样本中加入标准浓度的内标参考物,通过参考物的出峰时间和丰度对样本数据进行校正。脂质色谱出峰时间与不同类别脂质的极性、不饱和度及分子大小密切相关,同类脂质在保留时间维度具有明显规律,应用内标校正法可实现保留时间的有效校正。但该方法对内标的浓度和纯度要求较高,需研究人员依据样本中目标脂质信息,对内标参考物浓度等进行调整优化,以达到最好的效果,内标校正法适合样本量较少的脂质分析。该方法已被用于生物样本中磷脂、鞘脂信号的有效校正和鉴定分析[55-56]。
针对大样本量的脂质组学分析,运用保留时间校正算法来校正,能够有效降低数据处理的复杂性,缩短分析时间。色谱保留时间漂移通常包括线性和非线性两种。针对线性漂移,Nielsen等[57]提出了相关优化翘曲算法(Correlation optimised warping,COW),该算法先将色谱信号分成系列节段,再对每个节段进行线性翘曲校正,最后将校正后的节段重新组合,从而实现保留时间校正;但该方法易造成信号损失,需人为设置节段的长度和调整保留时间漂移相关参数来优化效果。Bylund等[58]在此基础上,应用更利于时间对齐的协方差作为校正效果函数,并引入了质谱m/z用以校正色谱信号。平行因子分析(Parallel factor analysis,PARAFAC)和主成分分析(Principal component analysis,PCA)对LC-MS信号校正的效果评价表明,该法比原COW算法更具实用性和选择性。值得注意的是,上述算法均需漂移时间小于邻近峰之间的保留时间差,才能实现准确有效的校正。
相比于色谱保留时间的线性漂移,因实验时间间隔差异和分析平台不同产生的色谱非线性保留时间漂移更为普遍[59]。Smith等[15]开发了基于非线性校准算法的工具包XCMS,该方法在提取离子色谱峰(Extracted ion base-peak chromatogram,EIBPC)的基础上,选择峰形完好的样本序列作为“临时标准”,再基于每组色谱信号的保留时间中值及其与样本序列的偏差进行校正,并结合低阶多项式的局部回归拟合算法排除异常值,实现保留时间的校正。Katajamaa等[60]开发了alignToMaster算法用以解决样本数据保留时间校正问题,该方法可自动识别生成峰序列,并参考邻近序列峰进行匹配校正,对于未识别的色谱峰,还可根据质谱碎片离子丰度实现自动补齐。但其缺点是校正准确性依赖于参考样本序列的构建,也需手动优化相关参数来提高校正的准确性。为实现更为准确和快速的非线性保留时间校正,Pluskal等[21]将RANSAC(Random sample consensus)算法应用于保留时间校正,该法可通过迭代来自动预测拟合模型的相关参数和异常值,随着迭代次数的增加,校正也更准确。集成上述非线性保留时间校正算法的XCMS和MZmine开源方法包,得到了众多研究者的认可和使用,被广泛用于靶向(Targeted)和非靶向(Untargeted)脂质组学数据的色谱保留时间校正。Masoodi等[61]应用XCMS成功对疾病细胞的LC-MS数据进行了保留时间校正,实现了复杂调控网络中的脂类生物活性介质(Lipid mediators)的综合分析。Cajka等[62]应用XCMS、MZmine等对血浆脂质组学数据进行处理,实现了包括溶血磷脂酰胆碱(Lysophosphatidylcholine,LPC)、磷脂酰胆碱(Phosphatidylcholine,PC)、磷脂酰乙醇胺(Phosphatidylethanolamine,PE)、溶血磷脂酰乙醇胺(Lysophosphatidylethanolamine,LPE)等12类495种脂质的有效校正和鉴定分析。
近年来,研究者们还基于核密度(Kernel model)、线性回归(Line regression)、自适应多维核函数(Tailored multidimensional kernel function)、分治算法(Divide-and-conquer algorithm)等模型开发了SIMA[63]、BIPACE[64]、PIRTA[65]等软件包,在逐步提高校正精度的基础上,实现了色谱保留时间的有效校正。
4 同位素信号解析
在脂质组学的质谱分析中,由于元素同位素的存在,脂质分子会产生稳定的同位素峰。在质谱谱图上表现为待测分子离子M之后低频、连续、稳定的信号峰[66-67]。一方面,同位素峰的存在会增加原始谱图中信号峰数量,产生重叠峰,干扰待测组分的有效识别;另一方面,同位素峰作为天然存在的现象,可从反面验证待测组分的存在,降低假阳性和假阴性识别率。研究表明,有效的同位素峰识别策略可作为解析复杂脂质组分的重要手段[68-70]。
现已建立的各种同位素信号识别算法,基于数学分离的手段,先利用数据函数模型对信号分布进行理论计算,再与实际同位素峰进行匹配筛选,实现同位素信号解析。相关化学计量学算法主要有多项式[71]、傅立叶变换卷积[72]、动态规整[73]等。基于多项式的经典同位素解析算法,通过不同原子的同位素质量和数目来构建高次多项式,实现分子同位素质量和概率分布的准确计算[70,74],但该方法拓展性较差,对于复杂分子的同位素计算会占用大量计算机资源,如计算分子C112H165N27O36S的所有同位素分布概率,需占用23.63 GB储存空间;在对大分子多项式系数阶乘计算中,还会出现溢出现象,导致同位素计算结果不准确;该方法繁琐费时,且难以实现生物大分子同位素的精确解析。Rockwood等[72,75]提出了快速预测同位素峰分布的傅立叶卷积算法,利用快速傅立叶转换(Fast fourier transform,FFT)将复杂理论同位素峰转换为呈整数等距分布的轮廓同位素峰(Profile mode isotopic variant)。该方法通过牺牲一定数据准确度来大幅减少计算量,解决了同位素信号解析耗时长、占用大量硬件资源的问题。其他研究者也尝试应用不同的算法来实现同位素信号的快速识别。如Mcilwain等[73]提出了基于动态规整(Dynamic programming)算法构建完整同位素峰的识别算法。Hussong等[25]将小波变换应用于同位素峰信号解析,提出了单尺度同位素小波变换算法(Single scale isotope wavelet transform),通过设置简单的阈值参数过滤识别同位素信号。Zhu等[76]提出了高分辨率质谱数据同位素快速识别算法(Accurate mass based spectral averaging isotope-pattern filtering,AMSA-IPF),该算法被编写成R包,用于同位素信号的自动处理。Liebisch等[68]基于同位素校正法,自编程了Excel宏程序,可在1.3 min内实现磷脂、鞘磷脂的同位素信号的快速分析。以上方法在一定程度改善了同位素信号识别准确度不高和耗时长的问题,但对于重叠的同位素信号,大多还不能进行有效的处理。Slawski等[77]将非负最小绝对偏差回归(Non-negative least absolute deviation regression,NLADR)算法应用于复杂同位素信号的识别,该法使用模拟的同位素模板库与原始谱图进行匹配,再应用自适应稳健阈值,对质谱信号进行筛选,进而实现重叠同位素峰的有效识别。与该法类似,Zeng等[78]开发了一种基于最小二乘的高分辨理论同位素分布算法(Least squares spectral resolution,LSSR)的Chrombox D方法包。该方法包先构建脂质加合物离子(如[M+H]+、[M+NH4]+、[M-H]-、[M-H2O]-等)的同位素谱图库,再应用该库对原始数据进行匹配,从而实现重叠同位素信号的快速分析。对鸡蛋、牛肝等生物样本的脂质分析结果显示,该算法可有效实现甘油酯、甘油磷脂、鞘磷脂等多类脂质的同位素信号识别,并可同时定性定量分析多种脂质组分。
针对不同的分析对象,研究人员需在同位素准确度和处理速度上予以考虑选择。对于小分子组分的定性定量研究,运用多项式能够准确预测和识别其同位素分布;对类似于蛋白、DNA等生物大分子的识别,选择基于傅立叶卷积、动态规整等快速识别算法,构建同位素轮廓谱,效率更高。
研究证明,整合了质荷比、保留时间、丰度、同位素和MS/MS数据库等多维信息的解析算法和软件包,在分析复杂脂质数据时准确度更高[18,21,23,46,79-82],表1列出了包含上述数据预处理流程和多维信息的开源数据处理软件的相关信息。除前文介绍的XCMS和MZmine软件包外,Hartler等[18]结合m/z、保留时间和同位素信息构建了3D算法,运用JAVA语言开发了脂质数据处理方法包Lipid Data Analyzer (LDA),并成功从C57BL雄鼠原代肝细胞脂滴中准确识别出了磷脂、甘油酯、鞘磷脂等657种信号峰。Tsugawa团队等[22]先应用RT、m/z等信息实现组分的初步筛选,再应用特有的MS2Dec(MS/MS deconvolution)算法提取到准确特征二级碎片信息用于二次验证,实现了脂质组分的准确鉴定。该算法已成功用于藻株脂质组学研究,并从藻株样本中准确鉴定了1 023种脂质成分。此外,中国科学院生物与化学交叉研究中心朱正江团队[83]基于m/z、碰撞界面CCS(Collision cross-section)、RT、MS/MS谱图四维信息,开发了LipidIMMS Analyzer脂质数据分析软件,结合该团队多维的脂质数据库,不仅能实现一般的脂质鉴定,还能有效区分生物样本中的脂质同分异构体。
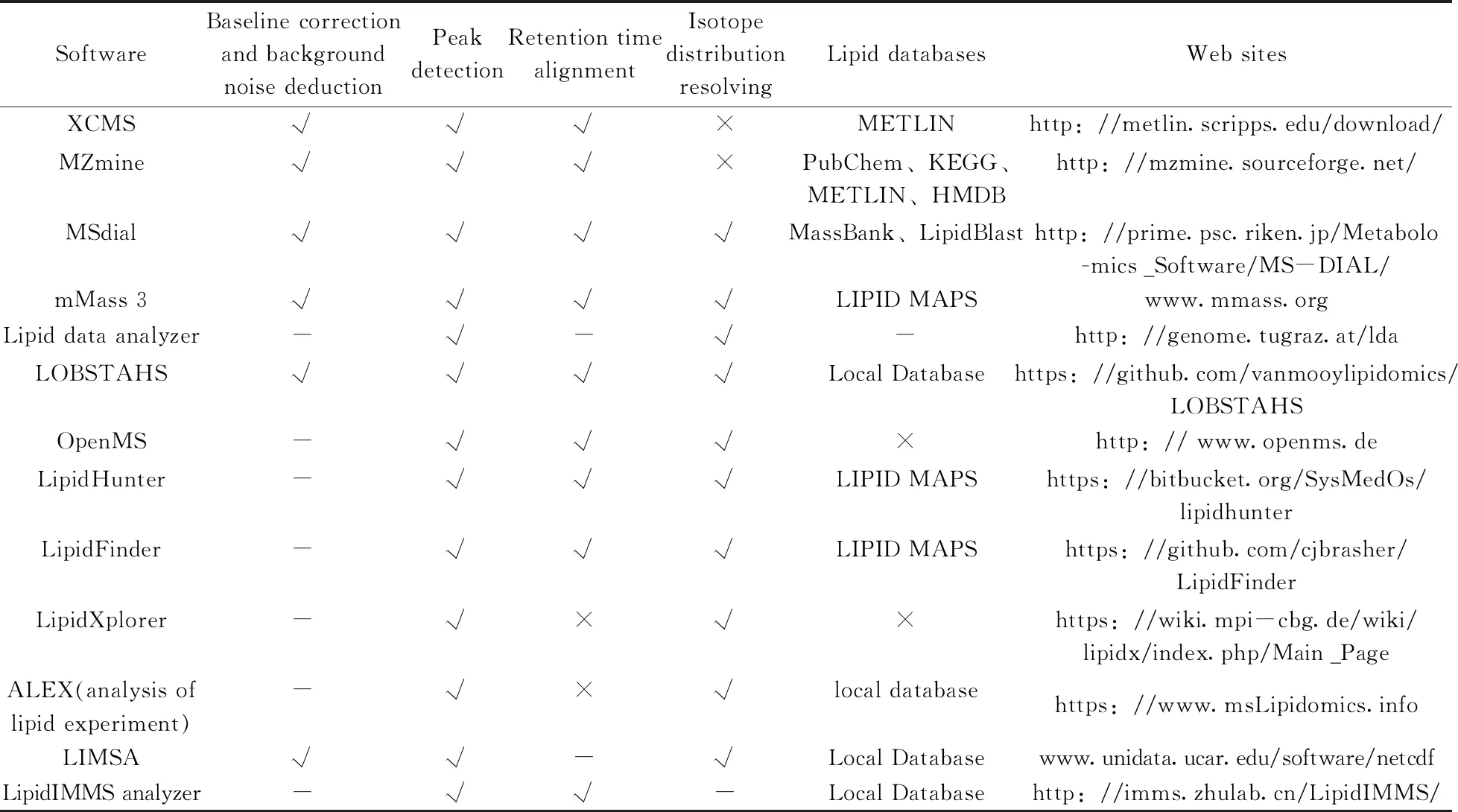
表1 脂质组学数据分析软件Table 1 Lipidomics data analysis softwares
“√” indicates that this function is available,“-”indicates that it is uncertain whether this function is available and “×” indicates that it does not have this function. “METLIN ”is the American metabolomics database, “PubChem” is the American small organic molecule database, “MassBank” is the database of Mass Spectrometry Society of Japan(MSSJ), and “Local database”is the software built-in database(“√”表示其有此功能,“-”表示据已有资料不确定是否有此功能,“×”表示无此功能;“METLIN”为美国代谢组学数据库,“PubChem”为美国有机活性小分子数据库,“MassBank”日本质谱学会质谱数据库,“Local Database”为软件内置本地数据库)
5 脂质组学数据统计分析
经过基线校正和背景噪声扣除、信号峰识别、色谱保留时间校正、同位素峰解析等数据预处理步骤,可实现脂质组分的定性定量分析。在此基础之上的统计学分析,可通过样本均值差异、总体分布或变量之间的相关性分析来挖掘样本的生物学意义和发现潜在的生物标记物。脂质组学统计分析主要包括单变量(Univariate significance tests)和多变量(Multivariate significance tests)差异分析。
单变量分析常用于比较简单样本的差异显著性,主要有t检验(t-test)、Wilcoxon假设检验、方差分析(Analysis of variance,ANOVA)等。t-test系列检验主要用于探究两组样本数据均值是否存在显著性差异;Wilcoxon假设检验可应用于单样本,检测样本浓度中位数与假设浓度中位数值是否存在区别,也可用于双样本,判断2个样本间的浓度中位数值差异是否显著;ANOVA分析类似于t-test检验,主要用于两组以上样本数据的均值差异比较,该法被广泛用于脂质组学的数据分析[84-86]。
在脂类研究中,多变量分析的应用更为广泛,包括主成分分析(Principal component analysis,PCA)、偏最小二乘法判别分析(Partial least squares discriminant analysis,PLS-DA)和多元方差分析(Multivariate analysis of variance,MANOVA)等。PCA分析是通过保留原始数据大部分相关信息的主成分变量来解释数据的变化。同时,主成分具有正交性,可有效避免数据的重复,再借助得分图和载荷图,可直接判断样本是否具有组间分类趋势和数据离群点。Shen等[87]将PCA用于鲈鱼、鲫鱼和草鱼3种鱼类内脏的磷脂差异分析。结果表明,PC和PE是3种鱼类内脏的主要脂质种类,其中磷脂酰丝氨酸(PS 18∶0/22∶6)、磷脂酰肌醇(PI 18∶0/20∶4)和磷脂酰肌醇(PI 18∶0/20∶5) 3种脂质还可作为鱼类物种分化的重要指标。PLS-DA分析是一种常见的样本分类方法,其将数据分为代表原始数据的矩阵X和代表分类信息的矩阵Y,通过选取矩阵中的典型相关因子来关联矩阵X和Y(Variables importance on projection,VIP)得分值,从而实现数据的分类。Sato等[88]将PLS-DA用于阿尔兹海默症患者的血浆磷脂酰丝氨酸(Phosphatidylserine,PS)数据的判别分析,依据VIP得分差异成功区分了阿尔兹海默症患者和健康对照组的血浆样本。其分析主要应用于涉及多种测量变量(如:多种脂质的浓度)的多变量因子主要作用和相互作用研究,根据不同的因子水平可实现组间的差异比较,其优点是可在减少无关测量变量数量,降低数据复杂度的条件下,实现样本数据的差异分析。在实际的脂质组学数据分析中,ANOVA、PCA和PLS-DA分析的结合使用更为普遍,广泛应用于脂质生物标记物挖掘、脂质的代谢通路研究[89-90]。
此外,一些其它的化学计量学方法也被应用于脂质组学的数据分析中,如随机森林(RF)、支持向量机(SVM)等。Cutler 等[91]提出了基于分类树的RF算法,分类树中的决策方法可应用于脂质组学数据的有效分类,这为脂质组学样本的分类和差异分析提供了一种新思路。SVM是一种基于核密度函数的非监督机器学习法,该方法先基于训练数据构造的超平面实现不同类别样本数据的分离[92]。但是以上两种方法的实际效果依赖于分类特征的选择,不同分类特征下,分析结果差异较大。脂质组学数据复杂、研究对象趋于多元,具体的统计分析问题,还需科研人员根据实际需求,结合不同化学计量学方法,灵活选择处理。
6 前景与展望
脂质组学数据处理具有数据复杂多维、分析流程复杂、分析过程耗时长等特点。目前,在脂质组学的数据处理方面已有较多算法和软件包,在一定程度上解决了数据处理耗时长、假阳性/假阴性识别等问题,降低了数据处理的难度。但仍然存在以下问题:①对于低浓度、低丰度的脂质信号,目前的算法和方法包仍然难以快速有效地识别。②针对大批量的原始数据,已有软件包和算法尚不能实现大规模自动化解析。③脂质相关数据库还不够完善,目前仅有40%~60%的脂类能够通过软件匹配得到有效识别,大量未知的脂类由于缺乏有效的信息,还无法进一步研究。④在脂质样本差异分析和生物标记物挖掘方面,研究人员需综合不同统计学方法的优点和不足,才能得到较为准确的结果。随着新型高维高通量分析仪器在脂质组学中的应用,数据结构将更为复杂,分析难度也会成倍增加。一方面,在现有算法和数据库进一步完善的基础上,整合了多种算法、数据库和统计分析等部分的完整数据自动解析平台将成为研究者进行数据解析的重要手段。另一方面,新的拟合模型和化学计量学方法如平行因子(Parallel factor analysis,PARAFAC)、多元曲线分率(Multivariate curve resolution alternating least squares,MCR-ALS)等[93-94]在数据解析中的应用,将为低丰度脂质准确定性定量、样本差异分析和生物标记物挖掘等提供新思路。未来,以化学计量学为手段,借助前沿计算机技术的数据自动化解析策略将推动脂质组学进一步发展。
猜你喜欢
昆明医科大学学报(2022年3期)2022-04-19
昆明医科大学学报(2021年4期)2021-07-23
中成药(2018年9期)2018-10-09
天然产物研究与开发(2018年2期)2018-04-04
中成药(2018年1期)2018-02-02
同位素(2018年1期)2018-01-18
中成药(2017年4期)2017-05-17
医学研究杂志(2015年11期)2015-06-10
同位素(2014年3期)2014-06-13
同位素(2014年2期)2014-04-16
