
荧光衍生化-磁分散固相萃取/高效液相色谱检测池塘水与蔬菜中3种挥发性异味物质
2020-05-08 13:40郑振佳董京磊孙鲁平朱树芸赵先恩
分析测试学报 2020年3期
陈 坤,张 鹏,王 妍,郑振佳,董京磊,孙鲁平, 朱树芸,赵先恩*
(1.曲阜师范大学 化学与化工学院,山东省绿色天然产物与医药中间体高校重点实验室,山东 曲阜 273165; 2.山东农业大学 食品科学与工程学院,山东 泰安 271018;3.龙大食品集团有限公司,山东 烟台 265200)
水体中的醇类挥发性异味物质(VOCs)主要包括2-甲基异莰醇(2-MIB)、土臭素(GSM)以及3-甲基-1-丁醇(3-MB)。随着人们对VOCs研究的不断深入,发现VOCs是产生水体污染的原因之一,且会影响饮用水、食品、养殖鱼类的味道和口感。VOCs在各种水体中含量极低,通常无法通过嗅觉判断其含量。因此,建立一种对VOCs进行高灵敏检测及质量控制的方法具有重要意义[1-3]。
已报道的VOCs等挥发性有机物的检测方法主要有气相色谱法(GC)、气相色谱-质谱法(GC-MS)以及分子印迹荧光位移法[4-8]。其中,GC法通常采用火焰光度检测器、电子捕获检测器和火焰离子检测器(GC-FID)对VOCs进行检测,但灵敏度较低且基质效应严重。GC-MS法报道较多,但检测灵敏度较低且仪器昂贵。分子印迹荧光位移法对VOCs的检测虽具有特异性,但灵敏度较低且制备过程繁琐。此外,VOCs的样品前处理方法较多,包括闭环捕集(CLSA)、液液微萃取(LLME)、吹扫捕集(P&T)、固相微萃取(SPME)等。但上述样品前处理技术对VOCs的研究结果往往难以令人满意,或操作较为复杂,或使用成本较高。目前虽有分析2-MIB和GSM的少量报道,但鲜有同时分析2-MIB、GSM与3-MB的研究。
对于食品和环境分析,衍生化结合前处理技术能简化样品的制备、降低基质效应、提高检测灵敏度,对食品安全监控和水体污染物的监测有重要意义[9]。磁分散固相萃取(MDSPE)技术可以选择性吸附小分子有机物的衍生化产物,且萃取后易于分离收集,操作快速简便[10]。氧化石墨烯具有超大的比表面积和良好的表面修饰性,可与化合物形成氢键、π-π堆积等作用[11]。使用磁性氧化石墨烯(MGO)进行MDSPE同时具有强吸附性和磁分离能力。因此,本研究建立了以6-碳酰氯左氧氟沙星(LFC-Cl)为衍生化试剂,MGO为MDSPE吸附剂的高效液相色谱-荧光检测(HPLC-FLD)分析方法。本方法实现了池塘水和蔬菜中前述3种VOCs的检测,具有灵敏度高、样品前处理简单、仪器普适性好等优势。
1 实验部分
1.1 仪器与试剂
Agilent 1260高效液相色谱仪(配备四元梯度泵、在线真空脱气机、荧光检测器、自动进样器、恒温柱温箱,美国Agilent公司),RE-2000B旋转蒸发仪(上海亚荣生化仪器厂),KQ2200E超声波清洗器(江苏昆山超声仪器有限公司),DF-2集热式磁力搅拌器(金坛市成辉仪器厂),真空干燥箱、高速万能粉碎机(北京市永光明医疗仪器有限公司),SC-06低速离心机(安徽中科中佳科学仪器有限公司),XW-80A旋涡混匀器(上海精科实业有限公司),SHA-C水浴恒温振荡器(金坛市金南仪器制造有限公司)。
左氧氟沙星(LFC,98.0%)、甲酸(色谱级,98.0%)、无水级1,2-二氯乙烷(99.8%)、4-二甲氨基吡啶(DMAP,99%)均购自阿拉丁公司,乙腈(99.9%,北京百灵威科技有限公司),三氯氧磷(POCl3,山东西亚化学股份有限公司),2-MIB(100 mg/L,德国Dr.Ehrenstorfer GmbH),GSM(10 ng/L,英国LGC集团),3-MB(99.0%,梯希爱化成工业发展有限公司),其他试剂均为分析纯。MGO参照本实验室方法合成[10]。
1.2 LFC-Cl的合成
LFC-Cl的合成参照文献[12-13]部分合成条件,并进行合成方案修改:向三颈烧瓶中加入1.0 g LFC和60 mL无水级1,2-二氯乙烷,用移液枪缓慢滴加2.2 mL POCl3,于85 ℃回流反应4 h。冷却后加入0.05 g活性炭,于85 ℃再次回流20 min。趁热过滤,除去活性炭,旋蒸除去溶剂得到LFC-Cl,置于真空烘箱中干燥8 h。干燥后的LFC-Cl产物可直接用于衍生化反应。
1.3 溶液配制
量取适宜体积的2-MIB、GSM、3-MB标准品,分别溶于乙腈得浓度为1.91×10-5mol/L的标准溶液,相应的低浓度溶液均由此标准溶液用乙腈稀释得到。取一定体积的3种标准溶液配成浓度为4.78×10-6mol/L的混合标准溶液。准确称取一定量DMAP溶于乙腈,得到浓度为0.20 mol/L的DMAP溶液。
1.4 衍生化反应
准确称取6 mg LFC-Cl于安瓿瓶中,加入250 μL乙腈、400 μL待测物混合标准溶液(或实际提取样品)、350 μL 0.05 mol/L的DMAP标准溶液,封口后,在60 ℃水浴中超声波辅助条件(超声波功率100 W,频率40 kHz)下衍生反应90 min。衍生化反应示意图见图1。衍生溶液待进行MDSPE过程。

图1 6-碳酰氯左氧氟沙星与含羟基挥发性异味物质的衍生化过程Fig.1 Derivatization reaction between LFC-Cl and hydroxyl-containing volatile organic compounds
1.5 MDSPE条件
MDSPE条件参考文献的部分实验方法[10,14],并修改如下:将20 mg MGO加入衍生溶液后,置于25 ℃水浴恒温振荡器中振荡20 min,在磁铁作用下分离弃去上清液,用1 mL乙腈洗涤3次,加入1 mL洗脱剂乙腈(含1%甲酸),在超声波辅助条件(超声波功率100 W,频率40 kHz)下解吸3 min,在磁铁作用下分离上清液,定容至1 mL,过0.45 μm有机相滤膜后稀释进样20 μL进行HPLC-FLD分析。
1.6 HPLC条件
色谱柱(CC):Agilent ZORBAX SB-C18柱(4.6 mm×150 mm,5 μm,美国Agilent公司)。流动相:A相为10%乙腈水溶液(含0.1%甲酸),B相为乙腈(含0.1%甲酸)。梯度洗脱程序(GE):0~6 min,2%B;6~10 min,2%~100%B;10~15 min,100%B。流速:1.0 mL/min,进样量:20 μL,柱温:30 ℃。荧光激发波长(λex)和发射波长(λem)分别为295、460 nm。
1.7 实际样品处理
实际样品处理参照文献的部分处理方法[7,12,15-16]。
1.7.1 池塘水处理量取50 mL池塘水于分液漏斗中,加入5.0 g 氯化钠固体,加入300 μL氯仿作萃取剂,萃取3次合并萃取液。用无水硫酸钠脱水干燥后定容至1 mL,待衍生。
1.7.2 蔬菜样品处理准确称取15.0 g绿色蔬菜(生菜、莴苣、葱)置于万能粉碎机打碎,收集后加入7.5 mL乙腈涡旋2 min,超声提取20 min。加入1.0 g氯化钠和4.0 g无水硫酸镁固体,涡旋5 min,以5 000 r/min 脱水15 min,待分层后准确吸取乙腈层,再次脱水后吸取乙腈层,待衍生化反应。
2 结果与讨论
2.1 HPLC条件的优化
实验对比了不同色谱柱(CC1:Agilent ZORBAX 300SB-C18柱(4.6 mm×250 mm,5 μm);CC2:Agilent ZORBAX SB-C18柱(4.6 mm×150 mm,5 μm);CC3:依利特 SinoChrom ODS-BP(4.6 mm×250 mm,5 μm);CC4:依利特HYPERSL C18(4.6 mm×100 mm,5 μm)),以及调整不同GE的分离效果,相同条件下平行测定3次(见图2)。对CC考察结果表明,采用CC2分离时3种VOCs衍生物的峰面积最大(图2A)。对GE考察结果表明,采用GE4时3种VOCs衍生物的峰面积最大(图2B),由于B相(含0.1%甲酸的乙腈)在0~6 min内已为2%,且能够实现3种VOCs衍生物的基线分离,因此采用GE4作为最佳梯度洗脱程序。采用Agilent 1260 HPLC-FLD仪器的在线荧光光谱扫描功能,确定3种VOCs衍生物HPLC-FLD的最佳激发和发射波长分别为295 nm和460 nm。最终确定了“1.6”所述色谱条件,3种VOCs衍生物在15 min内可实现基线分离(见图3)。


图3 3种挥发性异味物质衍生物的高效液相色谱图Fig.3 HPLC chromatogram of three derivatives of volatile organic compounds LFC:levofloxacin(左氧氟沙星);LFC-2-MIB:derivatives of 2-methylisoborneol(左氧氟沙星-2-甲基异莰醇衍生物);LFC-GSM: derivatives of geosmin(左氧氟沙星-土臭素衍生物);LFC-Cl: 6-carbonyl chloride levofloxacin(6-碳酰氯左氧氟沙星); LFC-3-MB:derivatives of 3-methyl-1-butanol(左氧氟沙星-3-甲基-1-丁醇衍生物)
2.2 衍生化条件的优化
实验同时考察了衍生化反应温度、反应时间、DMAP浓度以及衍生试剂用量对3种衍生物峰面积的影响,得到挥发性异味物质衍生化溶液后分别平行测定3次。
对反应温度和反应时间的考察结果表明:衍生物峰面积随着反应温度升高和反应时间的延长而增加,在60 ℃、90 min时峰面积达最大;继续升高反应温度或延长反应时间,衍生物的峰面积均减小(图4A和4B)。这可能是由于反应温度过高、反应时间过长使衍生产物发生分解所致。对DMAP浓度的考察结果表明:当DMAP浓度低于0.05 mol/L时,衍生物峰面积随其浓度的增大而增加;DMAP浓度为0.05 mol/L时衍生物的峰面积达最大;但当DMAP浓度大于0.05 mol/L后,衍生物的峰面积随其浓度的增加而减小(图4C)。这可能是由于催化剂DMAP的浓度增大导致反应产物在强碱性条件下分解。对衍生试剂用量的考察结果表明:衍生物的峰面积随衍生试剂用量的增加而增大,当衍生试剂用量为3种VOCs用量的1 500倍时,衍生物的峰面积最大,但继续增大衍生试剂用量,峰面积反而减小(图4D)。最终确定衍生化条件如“1.4”所述。
2.3 MDSPE条件的优化
实验同时考察了MGO吸附剂用量(10、15、20、25、30 mg)、萃取时间(10、15、20、25、30 min)、解吸时间(1、2、3、4、5 min)和解吸剂种类(均含1%甲酸的甲醇、乙醇、乙腈、丙酮)对萃取率的影响,得到异味物质衍生化溶液后分别平行测定3次。对吸附剂用量的考察结果表明:萃取率随着吸附剂用量的增加而增加,当吸附剂用量为20 mg时,萃取率最大;继续增加吸附剂用量,萃取率并无明显增大。对萃取时间的考察结果表明:萃取率随萃取时间的延长而增加,萃取时间为20 min时萃取率最大;继续延长反应时间,萃取率并无明显增大。对解吸时间的考察结果表明:萃取率随解吸时间的延长而增大,解吸时间为3 min时萃取率最大;继续延长解吸时间,萃取率并无明显增大。对解吸剂种类的考察结果表明:以乙腈(含1%甲酸)作解吸剂时的萃取率最大,这可能是不同解吸剂的极性、目标分子溶解度等存在较大差异,乙腈(含1%甲酸)能够更好地将异味物质衍生物从MGO中洗脱出来。最终确定MDSPE优化条件如“1.5”所述。



研究表明[11],MGO对有机化合物的吸附机理主要是吸附剂与被吸附分子形成氢键、π-π堆积、静电作用力、范德华力以及疏水作用等。本方法所使用的衍生试剂分子中存在可形成氢键的羰基、醚键、氟原子和氮原子等以及较大的共轭体系等结构,可与吸附剂形成上述作用力,因而产生良好的萃取效果。
2.4 方法评价
2.4.1 线性、检出限、定量下限与精密度在空白水样氯仿溶液中添加标准样品,按照衍生化方法和MDSPE过程进行处理,根据峰面积(Y)和实际进样浓度(X)进行线性回归,并分别以信噪比S/N>3和S/N>10得到3种衍生物的检出限(LOD)和定量下限(LOQ)。为便于与其他方法及文献中VOCs的检测结果进行对比,将3种VOCs衍生物的数据换算为VOCs的数据,结果见表1。结果表明,3种VOCs在一定质量浓度范围内线性关系良好,相关系数r≥0.987,LOD为0.020~0.95 ng/L,LOQ为0.10~3.3 ng/L。已有文献报道2-MIB和GSM的嗅味阈值约为10 ng/L[3],本方法测得3种VOCs的LOD和LOQ均低于VOCs的嗅味阈值,表明本方法可用于极低含量的VOCs样品检测。
在相同实验条件下,制备混合标准品衍生溶液,平行6次进样分析,得到3种VOCs衍生物保留时间和峰面积的相对标准偏差(RSD)均不大于1.7%(表1)。结果表明,本方法的精密度良好。
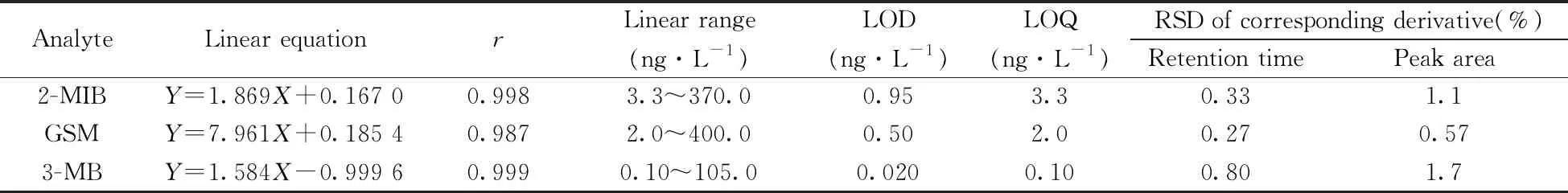
表1 3种异味物质的线性方程、相关系数、线性范围、检出限、定量下限与相对标准偏差(n=6)Table 1 Linear equations,correlation coefficients(r),linear ranges,LODs,LOQs and RSDs of three VOCs(n=6)
Y:peak area;X:injection concentration(ng/L)
2.4.2 与文献方法的比较与文献方法相比,本方法所用的衍生试剂LFC-Cl易于合成并首次被应用于VOCs检测。在检出限、操作难易程度和仪器普适性等方面的对比见表2。

表2 本方法与文献方法的比较Table 2 Comparisons of this method with the reported methods
*not reported(文献中未对此种物质进行检测)
结果显示,在检出限方面,本方法的检出限低于或与LLE/GC-MS[4,17]和LLME/GC-MS[7]法的检出限相近;而远低于SPE/GC法[6]和分子印迹荧光位移/GC-MS法[8]。这一方面是由于本方法使用的衍生试剂LFC-Cl具有较大的共轭体系,从而为高灵敏检测VOCs带来良好的效果。另一方面,MDSPE技术能够选择性地吸附小分子有机物的衍生化产物,起到富集和净化作用。在操作难易程度上,本方法使用的MDSPE与LLME、SPE等前处理方法相比无需特定设备,成本较低,且操作较简单;MGO的制备步骤比分子印迹材料少,且适用于多种异味物质衍生物的同时吸附。在仪器普适性方面,HPLC-FLD的普适性较好。
2.5 回收率与实际样品应用
以池塘水(取自曲阜市某池塘)和蔬菜(生菜、莴苣、葱,购自曲阜某菜市场)为实际样品,采用本方法进行前处理和实验,同时进行加标回收率实验。由表3可知,3种VOCs的回收率为76.8%~118%,能满足实际检测要求。实验发现,蔬菜样品中VOCs的含量较低,而池塘水中VOCs的含量较高。池塘水和蔬菜样品中3种VOCs衍生物的色谱图见图5。另外开展了对自来水、紫菜、海带等的适用性考察,测定结果的RSD均小于12%(n=5)。表明本方法的适用性良好,能够应用于各种实际样品中异味物质的检测。
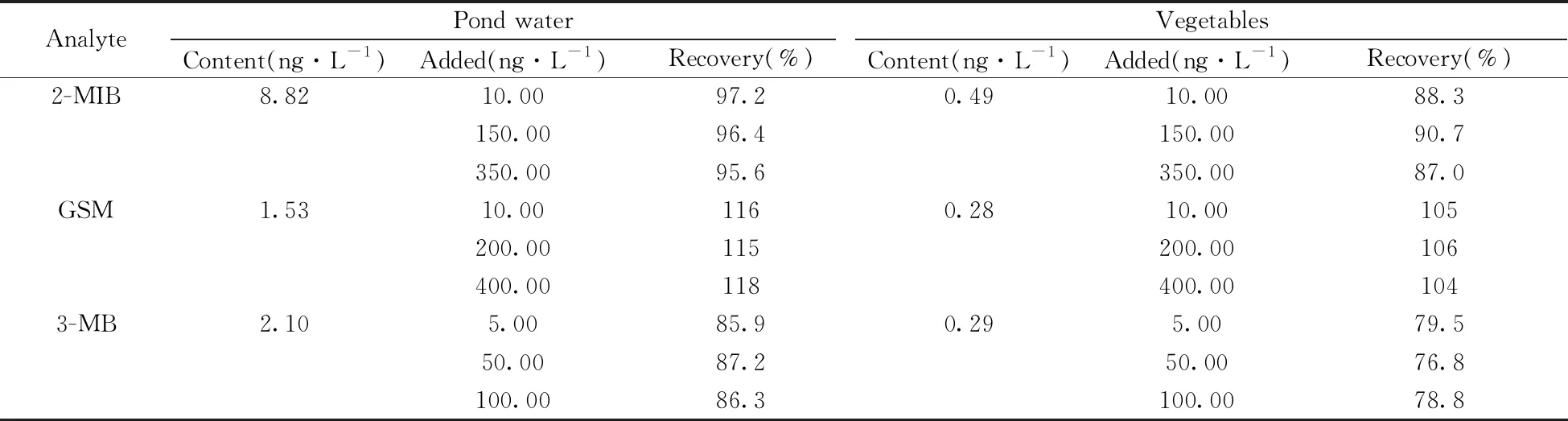
表3 两种实际样品中异味物质的含量及加标回收率(n=3)Table 3 Contents and recoveries of VOCs in two practical samples(n=3)
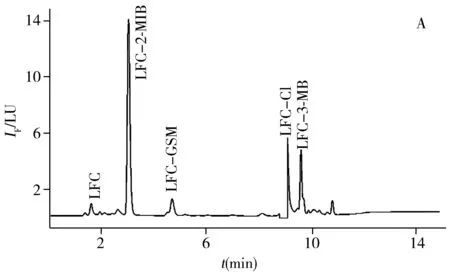
图5 池塘水样(A)与蔬菜样品(B)中3种异味物质衍生物的色谱图
Fig.5 Chromatograms of the three derivatives of VOCs in pond water(A) and vegetables(B)
3 结 论
本文利用衍生化技术结合磁固相萃取技术,建立了3种VOCs的HPLC-FLD分析方法。该方法具有灵敏度高、样品前处理简单、仪器普适性好等优势。实际检测结果表明,蔬菜中含有较少的VOCs,而池塘水中VOCs的含量相对较多。因此,需加强对各种湖泊池塘的管理和净化,以降低VOCs对人类健康的危害,减少对生态环境的破坏。
猜你喜欢
世界农药(2019年3期)2019-09-10
石油石化绿色低碳(2019年6期)2019-01-14
宠物世界·猫迷(2017年7期)2018-01-25
猪业科学(2018年8期)2018-01-22
中学生数理化·高二版(2016年3期)2016-12-26
天然产物研究与开发(2016年11期)2016-06-15
医学研究杂志(2015年4期)2015-06-10
家庭生活指南(2009年7期)2010-04-07
现代农业科技(2009年19期)2009-03-20
中学生数理化·八年级数学华师大版(2008年3期)2008-08-26
