
低氧诱导因子和脯氨酸羟化酶在骨发育和骨稳态中的作用
2020-05-05 13:00:28夏玉城陶树清
实用骨科杂志 2020年4期
夏玉城,陶树清
(哈尔滨医科大学附属第二医院,黑龙江 哈尔滨 150001)
低氧发生在骨骼发育的几个阶段。骨骼细胞通过低氧诱导转录因子(hypoxia inducible factors,HIF)诱导血管内皮生长因子(vascular endothelial growth factor,VEGF)的表达并促进糖酵解,增加氧气和营养物质的输送及代谢适应,以防止骨化中心内软骨细胞死亡,并促进成骨细胞的成骨作用。然而,在骨骼发育过程中必须避免过高的HIF水平,因为由此导致的代谢失调会使骨骼发育不良。最近的研究表明,HIF还以其他基因为靶点增加骨量:它通过增加骨保护素(osteoprotegrin,OPG)表达来减少破骨过程,通过抑制表观遗传骨硬化蛋白表达,使骨形成增加,骨吸收减少。此外,骨膜细胞中HIF信号的增加可促进原发性和转移性乳腺肿瘤的生长,并诱导促红细胞生成素(erythropoietin,EPO)的产生,导致红细胞增多症。最后,HIF通过EPO直接或间接诱导成纤维细胞生长因子23(fibroblast growth factor23,FGF23)的表达和加工,从而影响维生素D代谢和骨稳态。因此,骨骼细胞中的HIF信号不仅影响它们的行为,还能影响红细胞生成和骨稳态。
1 介 绍
在整个生命过程中,骨通过成骨细胞的骨生成和破骨细胞的骨吸收作用不断重塑,以维持其结构和功能的完整性[1-2]。骨骼细胞的功能不仅受激素、生长因子、神经系统信号和机械刺激的调控,还受局部微环境影响。氧气和营养物质对于正常的骨细胞功能来说非常重要,这一点已经得到广泛证实。虽然骨骼是一个高度血管化的器官[3],但骨微环境的特定区域氧含量却很低[4],这可能是由于毛细血管系统的组织血流量低以及造血骨髓细胞需氧量高导致的[5]。骨骼细胞通过感知低氧诱导转录因子(hypoxia inducible factors,HIF)的水平,从而对局部含氧量波动作出反应。HIF是细胞对低氧的反应,而脯氨酸羟化酶(prolyl hydroxylase,PHD)以氧依赖的方式调整HIF的水平。
转录因子HIF-1、HIF-2和HIF-3是低氧反应的中心介质,是由氧敏感的HIF-α亚基和结构性表达HIF-β组成的异二聚体亚单位[6-7]。在常氧环境中,HIF-α的半衰期小于5min,这种快速的常氧循环是以氧、二价铁和α酮戊二酸(α-ketoglutarate,α-KG)为底物,通过PHD使需氧区域内特定残基的羟基化,从而降解HIF的过程。已发现至少有三种PHD亚型(PHD1、PHD2和PHD3)羟基化HIF-α,它们是依赖α-KG的需氧双加氧酶家族的一种。羟基化促进HIF-α的泛素化,也是与肿瘤抑制蛋白(protein von hippel-lindau,pVHL)结合的关键,pVHL是E3泛素连接酶复合物的识别成分,以HIF-α为靶点进行蛋白水解降解。随着氧水平的降低,PHD羟基化的作用减弱,从而使HIF-α更加稳定[6-7]。这时HIF-α进入细胞核与HIF-β形成二聚体,并且在与转录共激活因子(如CBP/p300)结合后,进一步结合低氧反应基因启动子区域内的低氧反应元件(hypoxic response elements,HRE)。HIF信号的激活影响多种细胞和组织功能,但其主要是通过刺激血管生成来恢复降低的氧含量,并通过调节细胞代谢使细胞在低氧损伤中存活。
所有的骨骼细胞类型,包括软骨细胞、成骨细胞、骨细胞和破骨细胞,都表达了低氧信号通路的主要成分,而且越来越多的证据表明,在骨发育和骨稳态过程中调节低氧信号通路的重要性。
2 骨发育过程中的低氧、HIF和PHD
胚胎骨骼发育始于间充质干细胞(mesenchymal stem cells,MSCs)的聚集。虽然颅骨的扁平骨是由直接分化为成骨细胞(即膜内成骨)的MSCs发育而来,但其他骨则是由骨组织替代软骨而来[1-2],后一过程称为软骨内成骨,已有多项研究表明,低氧信号通路是一个关键的调节因子[8]。
2.1 HIF控制早期骨发育 通过软骨内成骨形成长骨是在多能MSCs聚集形成软骨细胞之前开始的[9]。在这些初始阶段,肢体血管系统经历了一个重塑过程,使聚集的MSCs缺乏血管,从而导致了低氧[10]。
肢体骨发育过程中,表达同源盒基因1(paired-related homeobox gene 1,Prx1)的细胞中HIF-1α的缺失不影响MSCs聚集,但延迟了MSCs分化为软骨细胞和软骨细胞的终末分化[10]。潜在的机制尚不完全清楚,但可能涉及HIF依赖的人体性别决定相关的高移动编码框基因9(SRY-related high mobility group-box gene9,SOX9)的调节,SOX9基因是肢体骨发育过程中的重要转录因子[10]。与HIF-1α相比,在缺乏HIF-2α的小鼠肢体骨中,只观察到骨骼发育的短暂延迟[11-12]。通过条件失活pVHL,进一步研究了与HIF-1α和HIF-2α的积累和低氧信号通路在肢体发育过程中的作用。表达Prx 1的肢体骨细胞中pVHL的缺失不影响MSCs向软骨细胞的转化,却影响软骨细胞的增殖、存活和终末分化,从而导致肢体缩短[13]。潜在的分子机制以及HIF氧传感器在这一过程中的作用仍然不清楚,需要进一步的研究。
综上所述,HIF-1α紧密调控早期肢体骨发育,包括MSCs的存活与软骨细胞的增殖和分化。
2.2 HIF与PHD的相互作用决定了软骨细胞在骨化中心的功能 MSCs聚集后,中央区域的MSCs分化为软骨细胞,软骨细胞开始增殖,形成软骨模板,为将来的成骨做准备。随后,中心区域的软骨细胞停止增殖,变得肥大,并与周围的骨母细胞共同产生血管内皮生长因子(vascular endothelial growth factor,VEGF),刺激血管向内生长[9]。成骨相关转录因子(Osterix,Osx)阳性骨母细胞与侵入血管一起进入软骨模板的过程[14],标志着初级骨化中心(primary ossification center,POC)发育的开始。低氧应激是否参与POC的形成,目前尚缺乏研究。另一方面,肥大软骨细胞表达的Runt相关转录因子2(Runt-related transcription factor 2,Runx2)可以稳定HIF-1α,从而以不依赖低氧的方式驱动血管向内生长[15]。
POC的形成,限制了软骨细胞在长骨两端的生长。在骨化中心内细胞向纵轴方向发展,增殖的软骨细胞位于骨末端,肥大的软骨细胞位于骨干处。随着胎儿骨化中心在无血管情况下的扩张,中心缺氧更加严重,因此HIF-1α是软骨细胞存活的关键调控因子[16]。事实上,表达Ⅱ型胶原(collagen type 2,COL2)的软骨细胞中HIF-1α的条件性缺失可导致严重的细胞死亡,这在中心区域更加显著[16]。由于骨化中心内缺乏血管,低氧激活软骨细胞中的HIF通路,通过诱导血管生成改善氧和营养供应。表达于增生和肥大软骨细胞内的VEGF,是最有效的内皮细胞有丝分裂原[17-18],同时也是HIF的直接靶向基因[19]。在小鼠软骨细胞中条件性缺失VEGF或联合缺失VEGF120和VEGF164导致细胞死亡[20-21]。然而,上述细胞的死亡没有HIF-1α条件敲除的细胞那么明显,这表明HIF至少在一定程度上通过VEGF信号和随后的血管生成来调节细胞生存。事实上,转基因VEGF164的表达[16]并不能完全挽救HIF-1α突变小鼠软骨细胞的死亡,这说明依赖HIF的细胞可以自主调控细胞存活。HIF-1α可能通过调节糖酵解酶过程中磷酸甘油酸激酶-1的表达来调节软骨细胞的代谢[16],从而在低氧情况下存活。此外,HIF-1α还控制软骨细胞的增殖以及基质的合成和修饰[22-23]。然而,增殖效应主要发生在骨化中心边缘,且HIF-1α仅适度表达,这可能是中心细胞死亡的代偿性反应。
骨化中心低氧信号通路的局部激活提示HIF水平必须在其他无血管区域被严格调控。最近研究证明了PHD2调控的HIF-1α失活对于避免代谢诱导的骨骼发育不良是必要的。事实上,PHD2缺失导致HIF-1α不能被激活,会降低葡萄糖氧化,从而限制软骨细胞的增殖和骨的纵向生长。此外,谷氨酰胺衍生物与α-KG的增加可以使胶原过度修饰和骨量增加[24]。在表达胶原蛋白的软骨细胞中,pVHL的缺失降低了细胞增殖,增加了骨化中心内基质的沉积[23],进一步支持了维持适当的HIF水平的调节机制对正常骨发育的重要作用。
因此,HIF-1α是骨化中心内软骨细胞的生存因子。虽然无血管环境,但是HIF的水平必须严格控制,以避免代谢诱导的骨骼发育不良。
3 通过HIF和PHD维持出生后骨稳态
虽然成骨的外观不会发生变化,但通过破骨细胞介导的骨吸收和成骨细胞介导的骨形成不断更新[1-2]。与骨化中心相比,骨髓高度的血管化,这些血管为骨细胞提供氧气和营养物质,同时也为造血干细胞和骨母细胞提供生存环境[3]。尽管骨是高度血管化的器官,但骨微环境的特定区域含氧量较低[4]。成骨细胞的低氧信号通路参与了血管生成以及成骨和血管生成过程之间的耦合。骨母细胞或成熟成骨细胞中HIF-1α条件性缺失导致骨体积和血管数量减少,血管数量减少是由于局部VEGF生成下调所致[25-26]。然而通过pVHL或PHD在骨膜细胞中缺失而激活HIF通路则结果相反。血管密度的变化是一个重要的驱动因素,但骨膜细胞的HIF信号也控制骨稳态和骨细胞功能,且不依赖于血管生成的增加[27]。最近的研究显示,首先在出生后小鼠的Osx阳性骨母细胞中,稳定的HIF-1α使小梁骨数量增加,这与骨血管的增加有关。然而,骨量增加并不继发于血管生成的增加,而是依赖于骨母细胞糖酵解的上调,但是VEGF的条件性缺失,完全逆转了HIF-1α依赖的骨形成[28-29]。糖酵解增加究竟如何促进成骨细胞分化和骨形成仍然未知。其次,成骨细胞低氧信号通路的激活也会影响与其他骨骼细胞的相互作用,从而调节HIF依赖的骨稳态。实际上,在Osx阳性祖细胞中联合灭活PHD2和PHD 3可通过减少骨保护素(osteoclastogenesis inhibitory factor,OPG)及HIF驱动的破骨活动,从而增加骨量[30]。此外,在骨细胞中HIF激活后,抑制了骨硬化蛋白的表观遗传,使骨形成增加、骨吸收减少,骨量增加[27,31]。最后,骨祖细胞中HIF信号的增强导致骨量增加,同时增加造血干细胞和红细胞系的选择性增殖。事实上,pVHL基因消融及PHD1/2/3基因联合缺失,或用药物抑制PHD,可以增加造血干细胞(hepatic stellate cells,HSCs)的数量,从而增加经过高强度辐射的小鼠细胞的存活数量。此外,突变小鼠出现了红细胞增多症,这是由HIF增加成骨细胞促红细胞生成素(erythropoietin,EPO)所致。值得注意的是,该模型中造血干细胞的增加可能部分依赖于血管生成,因为骨母细胞中HIF信号的激活导致血管内皮生长因子介导的骨髓血管系统的生长[28],而骨髓血管系统是HSCs存在的关键部位[32](见图1)。
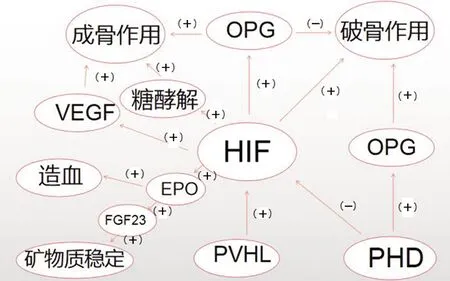
图1 HIF和PHD维持出生后骨稳态示意图
最近的数据表明,成骨细胞中HIF信号的增加可能直接或间接地诱导EPO生成,从而调节成纤维细胞生长因子23(fibroblast growth factor 23,FGF23)水平。正常情况下,FGF23主要由骨细胞产生,这种激素在肾脏磷酸盐的重吸收和产生1,25-二羟维生素D3时减少。后者是由1a-羟化酶表达减少和24-羟化酶水平升高引起的。与矿物质有关的因素是FGF23的主要调节因子,如磷酸盐、1,25-二羟维生素D3和甲状旁腺激素为正向调节因子[33]。然而,人们逐渐证实了包括HIF和EPO在内的非矿物因子调节FGF23产生及裂解。只有全段成纤维细胞生长因子23(intact fibroblast growth factor23,iFGF23)才具有生物活性,而裂解片段的作用尚不完全清楚。HIF-1α可能由炎症或缺铁引起[34],可与FGF23启动子结合,增加其在成骨细胞中的表达[35]。X-连锁低磷酸盐血症是一种具有高生物活性的FGF23水平的疾病。在模拟这种疾病的羟脯胺酸小鼠模型中,HIF-1α信号在成熟成骨细胞中的失活既没有改变血清中iFGF23水平,也没有改变骨稳态,这表明在本模型中成骨细胞HIF和FGF23之间没有联系[36]。然而,HIF信号在其他骨膜细胞和非骨细胞中的作用不能排除。除了直接转录调控外,HIF-1α还可能通过促进EPO的产生而增加FGF23水平。EPO增加了FGF23的转录和FGF23的裂解,因为体内FGF23水平与iFGF23水平的增加不成比例[37-39]。在一些研究中,血清FGF23水平的升高与血清磷酸盐或1,25-二羟维生素D3的水平降低有关,这反过来可能改变骨骼的稳态[37]。EPO通过靶向骨髓中的非骨细胞,包括红细胞系和造血干细胞,对FGF23表达产生影响[37-39]。然而,EPO调控FGF23转录和裂解的机制尚未完全阐明。在人类中观察到EPO和FGF23水平之间也存在相似的关联,FGF23总水平的增加大于全段FGF23水平的增加[37-39]。FGF23片段的增加是否具有病理生理学意义仍有待阐明。综上所述骨骼功能正常需要HIF信号转导,通过代谢适应直接调节成骨细胞功能,并通过局部分泌VEGF、OPG、骨硬化蛋白和EPO间接影响骨髓内皮细胞、破骨细胞和造血细胞,而EPO又可能改变FGF23水平及骨稳态。
几项研究表明,HIF对正常骨发育和骨稳态至关重要,保护骨骼细胞免受低氧的损害[8]。血管生成的增加和代谢的适应是促进机制,但缺乏对HIF驱动成骨细胞代谢的全面认识。在软骨细胞中,大量的HIF影响代谢平衡,并诱导骨骼发育不良。除了调节骨骼发育和骨量,最近在骨骼细胞中还发现了一些新的HIF的靶点,这些靶点可以影响其他类型的细胞,包括减少破骨发生的OPG和促进红细胞生成的EPO。EPO除了在调节造血细胞方面的作用外,还可能增加FGF23的生成和裂解,但其机制仍有待确定。此外,成骨性HIF在EPO-FGF23通路中的作用还需要进一步用成骨细胞和/或肾特异性灭活HIF或PHD的小鼠模型进行体内验证。这些发现对于PHD抑制剂治疗慢性肾病性贫血、骨发育不良及骨质疏松的临床转化具有重要意义。
猜你喜欢
中国骨质疏松杂志(2024年2期)2024-03-19 09:30:14
中国骨质疏松杂志(2021年9期)2021-10-08 10:07:40
天津医科大学学报(2021年3期)2021-07-21 09:03:42
中国临床医学(2019年3期)2019-01-04 09:12:32
安徽医科大学学报(2016年12期)2017-01-15 14:21:53
国外医药(抗生素分册)(2016年5期)2016-07-12 14:25:34
中国民族医药杂志(2016年6期)2016-05-09 08:52:52
中国医科大学学报(2015年10期)2015-03-01 02:09:58
实验动物与比较医学(2014年5期)2014-02-28 14:53:12
中国医学科学院学报(2012年3期)2012-03-25 13:58:48
