
酿酒酵母异源合成己二酸
2020-05-04 07:56:48张熙李国辉周胜虎毛银赵运英邓禹
食品与发酵工业 2020年7期
张熙,李国辉,周胜虎,毛银,赵运英*,邓禹*
1(粮食发酵工艺与技术国家工程实验室(江南大学),江苏 无锡,214122) 2(江南大学 生物工程学院,江苏 无锡,214122)
己二酸是一种具有高附加值的化学品,主要被应用于合成尼龙6,6的前体,后者是一种性能优良的纺织材料,可运用在防火系统、宇航制品等先进领域[1]。尽管全球经济不景气,己二酸的市场规模保持了每年约3.7%~5%的复合增长率,预计在2019年达到75亿美元[2]。目前,己二酸的生产主要以石油来源的环己醇和环己酮为原料[3-4]。在生产过程中,反应条件十分剧烈,并随着硝酸的氧化作用释放出大量的氮氧化物,产生巨大的环境压力[5]。另外,面对日益枯竭的石油资源,己二酸替代生产方案的挖掘势在必行[6]。2004年,美国能源部发布了一份包含10种“最具价值的生物基化学品名单”,己二酸位列其中[7]。
近年来,微生物代谢工程的发展使低成本、高效率的生物转化逐渐获得青睐。在2015年,己二酸首次在重组大肠杆菌中合成,主要使用了内源β-ketoadipyl-CoA thiolase(PaaJ)、来源于Ralstoniaeutropha的 3-hydroxyacyl-CoA reductase(PaaH1)和enoyl-CoA hydratase(Ech)、来源于Euglenagracilis的trans-enoyl-CoA reductase(Ter)、来源于Clostridiumacetobutylicum的 butyryl kinase(Buk1)和phosphate butyryltransferase(Ptb)[8]。然而,这条基于“逆向β-氧化”设计的途径,产量只有0.6 mg/L;通过一系列基因敲除(ΔldhA, ΔpoxB, Δpta, ΔadhE, ΔsucD),己二酸的产量被提高到了2.5 g/L[9]。另外,基于其他途径如逆β-then-ω氧化途径、2氧代庚二酸途径、2氧代己二酸途径、赖氨酸途径等均被尝试,并没有得到良好的效果[10-11]。
邓禹等[12-13]在T.fuscaB6 突变株中发现了一条可以天然合成己二酸的合成途径,被命名为Tfu己二酸逆降解途径。系统分析结果表明,己二酸是由乙酰辅酶A和琥珀酰辅酶A缩合,并经历5步反应得到——β-ketothiolase(Tfu_0875)、3-hydroxyacyl-CoA dehydrogenase(Tfu_2399)、3-hydroxyadipyl-CoA dehydrogenase(Tfu_0067)、5-carboxy-2-pentenoyl-CoA reductase(Tfu_1647)和adipyl-CoA synthetase(Tfu_2576-7)(图1)。但考虑到T.fusca遗传背景不清晰、基因操作工具少,该课题组将Tfu己二酸逆降解途径导入大肠杆菌,并将乳酸、丁酸和乙酸等途径进行敲除或弱化,以甘油为碳源实现了68 g/L己二酸的产量,为目前已报道的最高值[14]。

图1 Tfu己二酸逆降解代谢途径
大肠杆菌等细菌在生产有机酸时需要将pH控制在中性,需要补加大量的碱对发酵环境进行中和,极大地增加了发酵生产成本;另外,在高底物或产物浓度环境中,大肠杆菌等细菌由于其细胞壁结构的原因,耐受性较差,因此在二元羧酸的生产中,酿酒酵母成为了一个更优良的选项。酿酒酵母是一种具有高抗逆性、高酸耐受性和高底物耐受性的真核模式菌株,并以其相对清楚的基因背景,成为有机酸发酵生产中常用宿主。以苹果酸和丁二酸为代表的二元羧酸均以重组酿酒酵母获得了较高的产量,分别达到 59和 43 g/L[15-16]。
近年来,在葡萄糖二酸、黏糠酸等高附加值六碳二元羧酸的研究中,酵母也体现出了更加明显的优势:本课题组在酿酒酵母平台中构建葡萄糖二酸生物合成途径,实现了6 g/L 的目标物产量[17];LEAVITT等[18]在酿酒酵母平台中实现了2.1 g/L 黏糠酸的产量。有关己二酸在酿酒酵母平台的生产报道较少,仅有一个课题组报道:该课题组在LEAVITT等研究的黏糠酸生产菌株的基础上,加入了微生物 enoate reductases(ERs),并通过发酵优化由葡萄糖实现了2.59 mg/L 己二酸的产量[19]。基于目前酿酒酵母平台较低的己二酸产量及细菌类平台的耐酸性问题,本研究将Tfu己二酸逆降解途径首次导入酿酒酵母平台,探索该途径在酿酒酵母体系中的应用潜力。
1 材料和方法
1.1 材料
1.1.1 菌株和质粒
本研究中使用的菌株和质粒见表1。

表1 菌株和质粒
1.1.2 培养基
YPD培养基:20 g/L葡萄糖,20 g/L蛋白胨,10 g/L酵母抽提物。若需要固体培养基,添加20 g/L 琼脂粉。
LB培养基:10 g/L蛋白胨,5 g/L酵母抽提物, 10 g/L NaCl。若需要固体培养基,添加20 g/L 琼脂粉。
SD培养基:20 g/L葡萄糖,5 g/L(NH4)2SO4,1.7 g/L YNB,加入适量必需氨基酸。若需要固体培养基,添加20 g/L 琼脂粉。若需要缺陷型培养基,则再去掉某种缺陷标记氨基酸的添加。
1.1.3 仪器和试剂
PCR仪,美国Bio-red;高效液相色谱仪,美国安捷伦;MALDI SYNAPT Q-TOF MS液相-质谱仪,美国Waters;5 L发酵罐,中国迪必尔;高速冷冻离心机,美国赛默飞;电泳仪,中国六一。
DNA聚合酶Prime Star Max,日本宝生物;DNA连接酶,中国生工;Gibson 组装试剂,美国NEB;限制性内切酶,日本宝生物;DNA纯化试剂盒及质粒小量抽提试剂盒,中国生工。
1.2 实验方法
1.2.1 仪器和试剂
Tfu己二酸逆降解途径共涉及5步反应,6个基因,分别为Tfu_0875、Tfu_2399、Tfu_0067、Tfu_1647、Tfu_2576、Tfu_2577。从上获取到基因序列,并在http://sg.idtdna.com/CodonOpt网站上进行密码子优化,送往苏州泓讯进行基因合成。基因合成在3个T载体上,得到载体pUCmT-0875-2399、pUCmT-0067-1647和pUCmT-2576-2577,通过引物0875-F/R、2399-F/R、0067-F/R、1647-F/R、2576-F/R、2577-F/R进行扩增后得到基因片段T-0875、T-2399、T-0067、T-1647、T-2576和T-2577,纯化后进行下一步实验。本研究中使用的引物序列见表2。
1.2.2 表达Tfu途径酶质粒的构建
按顺序分别将6个途径酶基因搭配不同的营养缺陷型标记、启动子和终止子,得到穿梭质粒P1-V1、P2-V1和P3-V1。以酿酒酵母BY4741的gDNA为模板,用引物CN9847-1-AF/R、CN9847-1-BF/R、CN9847-1-DF/R、CN9847-1-EF/R、CN9847-1-GF/R分别扩增基因片段Tcyc1-1、Ttef1-1、Ptef1-1、Pgpd1-1、Ttdh2-1,与基因片段T-0875、T-2399按照图2-a顺序进行Gibson组装,各个片段全长连接至T载体,得到中间构建pUCmT-Tfu_0875-Tfu_2399,命名为P1;将中间构建体pUCmT-Tfu_0875-Tfu_2399与质粒pRS423(His)使用XhoI与SacI双酶切,再用T4DNA连接酶连接得到重组质粒pRS423-Tfu_0875-Tfu_2399,命名为P1-V1(图2-b)。以酿酒酵母BY4741的gDNA为模板,用引物CN9847-2-AF/R、CN9847-2-BF/R、CN9847-2-DF/R、CN9847-2-EF/R、CN9847-2-GF/R分别扩增基因片段Ttdh2-2、Tadh1-2、Padh1-2、Ppgk1-2、Tpgk1-2,与基因片段T-0067、T-1647按照图2-a顺序进行Gibson组装,各个片段全长连接至T载体,得到中间构建体pUCmT-Tfu_0067-Tfu_1647,命名为P2;将中间构建体pUCmT-Tfu_0067-Tfu_1647与质粒pHAC181(Leu)使用NdeI与EcoR I双酶切,再用T4DNA连接酶连接得到重组质粒pHAC181-Tfu_0067-Tfu_1647,命名为P2-V1(图2-b)。以获得的酿酒酵母BY4741的gDNA为模板,用引物CN9847-3-AF/R、CN9847-3-BF/R、CN9847-3-DF/R、CN9847-3-EF/R、CN9847-3-GF/R分别扩增基因片段Tpgk1-3、Ttpi1-3、Ptpi1-3、Ptdh3-3、Tfab1-3,与基因片段T-2576、T-2577按照图2-a顺序进行Gibson组装,各个片段全长连接至T载体,得到中间构建体pUCmT-Tfu_2576-Tfu_2577,命名为P3;将中间构建体pUCmT-Tfu_2576-Tfu_2577与质粒Y42(Ura3)使用BamH I与EcoR I双酶切,再用T4DNA连接酶连接得到重组质粒Y42-Tfu_2576-Tfu_2577,命名为P3-V1(图2-b)。

表2 引物序列
注:下划线表示同源臂序列

a-途径构建;b-质粒图谱
1.2.3 酿酒酵母LSC1基因的敲除
以pUG6质粒为模板,用引物ΔLSC1-pUG6-F/R扩增LoxP-KanMX-LoxP片段;以酿酒酵母gDNA模板,用引物LSC1-Upstream-F/R和LSC1-Downstream-F/R分别扩增LSC1基因上下游500 bp的片段。以OE-PCR方法将上述片段融合,得到LSC1基因敲除框。
以融合得到的敲除框为模板,用引物LSC1-Upstream-F和LSC1-Downstream-R进行PCR扩增,得到高浓度敲除框,纯化后转化酿酒酵母BY4741进行基因敲除筛选。具体的转化方法如下:将酿酒酵母BY4741活化后,在1 mL YPD液体培养基中培养至对数期(OD600值为0.6~1.0),5 000 r/min离心得到菌体,用双蒸水洗2次,并用0.1 mol/L LiAc洗2次,弃上清液,按顺序加入240 μL体积分数50% PEG、36 μL 1 mol/L LiAc、20 μL ssDNA(10 mg/mL, 沸水浴5 min)、4 μg 敲除框DNA。在振荡器混合30 s后置于30 ℃金属浴1 h,转移至42 ℃热激40 min,5 000 r/min离心,用双蒸水洗2次后涂布YPD-G418平板,培养2~3 d。挑取阳性转化子提取基因组,用引物ΔLSC1-verify-F/R进行验证。得到的正确菌株命名为BY4741ΔLSC1。
1.2.4 Tfu途径酿酒酵母菌株的构建
将构建得到的穿梭质粒P1-V1、P2-V1和P3-V1转化至BY4741野生型和BY4741ΔLSC1菌株中。具体转化方法如下:将酿酒酵母BY4741或BY4741ΔLSC1活化后,在1 mL YPD液体培养基中培养过夜,5 000 r/min离心得到菌体,用双蒸水洗2次,弃上清液,在体系中按顺序加入160 μL体积分数50% PEG、50 μL 1 mol/L LiAc、10 μL ssDNA(10 mg/mL, 沸水浴5 min)、20 μL体积分数100% DTT,待转化质粒各2 μg。在振荡器混合30 s后置于42 ℃热激45 min,5 000 r/min离心,用双蒸水洗2次后涂布SD-Leu-His-Ura平板,培养2~3 d。得到的菌株分别命名为AA-1和AA-3。
1.2.5 Tfu途径酿酒酵母菌株的发酵和样品处理
将构建得到的菌株AA-1和AA-3在SD-Leu-His-Ura平板上活化,挑取单菌落转接至SD-Leu-His-Ura液体培养基培养过夜,按照最终OD600=0.5的接种量接种至YPD液体培养基,30 ℃、250 r/min条件下发酵。每12 h 取样0.5 mL,样品经5 000 r/min离心,得到发酵上清液;菌体用双蒸水洗2次后补齐至EP管0.5 mL刻度线,加入与菌体量相当的玻璃珠,在振荡器上振荡1 min,冰浴1 min,循环6次,12 000 r/min离心10 min,上清液即为菌体破碎液。将发酵上清液和菌体破碎液进入后续的高效液相色谱仪或液质联用仪检测。
1.2.6 样品的分析与检测
液相色谱分析方法:将处理后的发酵样品,用0.22 μm的滤膜过滤,上HPX-87H有机酸柱(Bio-red),流动相为5 mmol/L H2SO4,流速 0.6 mL/min,柱温30 ℃。
液相质谱分析方法:将处理后的发酵样品,用0.22 μm的滤膜过滤。质谱柱为Waters Acquity UPLC BEH C18柱(2.1 mm×50 mm),检测器为Waters Acquity PDA detector(200~400 nm)。流动相A,体积分数0.1%甲酸;流动相B,体积分数100% MeOH;流速,0.3 mL/min,梯度洗脱,柱温,45 ℃。质谱检测条件:等度洗脱;电喷雾电离正离子源(ESI+);毛细管电压3.5 kV;锥孔电压30 V;离子源温度100 ℃;去溶剂气温度400 ℃;去溶剂气流速700 L/h;锥孔气流速50 L/h;电离能量6 eV;MS质量范围(m/z)20~2 000;检测器电压1 800 V。
2 结果与分析
2.1 表达Tfu途径的菌株构建
通过外源基因和不同启动子、终止子搭配的方式构建得到的3个重组穿梭质粒,使用验证引物pRS423-veri-F/R、pHAC181-veri-F/R和Y42-veri-F/R分别进行PCR,得到正确的条带如图3所示,PCR产物大小依次为4 936、4 469和4 752 bp。各外源基因均搭配酿酒酵母组常用的成型启动子,如图2-a所示。P1-V1、P2-V1、P3-V1质粒均为中高拷贝质粒,可以确保基因的表达量(图2-b)。将上述经转化酿酒酵母BY4741得到AA-1菌株。
LSC1基因编码催化琥珀酰辅酶A转化为琥珀酸的酶关键亚基[20]。通过LSC1基因的敲除,从理论上来讲,可以使Tfu途径的前体物质——琥珀酰辅酶A得以保留;但切断的TCA循环可能导致菌体生长和代谢的不正常。使用BY4741ΔLSC1菌株基因组为模板进行验证PCR得到阳性结果,得到正确的BY4741LSC1基因敲除株(图4)。该菌株经P1-V1、P2-V1、P3-V1质粒的转化,得到了AA-3菌株(图2-b)。

M-5 000 bp marker;1-P1-V1验证;2-P2-V1验证;3-P3-V1验证

M-5 000 bp marker;1,2-正确敲除株;3-未正确敲除株;4-空白对照株
2.2 Tfu途径菌株的发酵验证
本研究将Tfu己二酸逆降解途径完整地表达在酿酒酵母AA-1及其LSC1基因敲除株AA-3中,以BY4741野生型菌株作为对照,经过84 h的发酵,分别检测了生物量、己二酸和乙醇的产量变化(图5)。

图5 菌株AA-1、AA-3和BY4741在YPD培养基发酵中生物量的变化趋势
在生物量变化方面,48 h以前,野生型菌株生长最快,在36 h已接近稳定期,生物量增长放缓;AA-1菌株生长稍显缓慢,但在48 h生物量达到了11 g/L左右,与野生型相似;AA-3菌株生长缓慢,24 h之后才进入对数期,48 h进入稳定期。但在48 h以后,各菌株生长减缓,生物量峰值接近,为10~12 g/L。可能的原因是:1)AA-1菌株含有3个重组质粒,代谢负担较重,因此生长比野生型稍显迟缓;2)AA-3为TCA循环切断菌株,减缓了有氧呼吸速率,因此生物量生长缓慢;3)3株菌最终的生物量达到一致,说明了总碳源氮源相同的情况下,菌体生物量的极值趋于相同。
如图6所示,在己二酸产量方面,AA-1菌株在24 h 达到了1.12 mg/L,48 h上升到了最大值3.39 mg/L,但在72 h 时发生了下降;令人意外的是AA-3在48 h仅仅检测到0.33 mg/L己二酸产量,而24 h 和48 h 均未检测到;野生型没有检测到己二酸的生成。己二酸产量在达到最大值后发生下降,可能的原因是己二酸通过该途径发生了部分降解,该现象与大肠杆菌的报道一致。而AA-3的己二酸产量极少,可能是由于LSC1基因的敲除导致了TCA循环的流量中断,没有更多的碳源向积累琥珀酰辅酶A的方向流动。

图6 菌株AA-1、AA-3和BY4741在YPD培养基发酵中己二酸的产量
如图7所示,在主要副产物乙醇的产量方面,AA-1和野生型菌株呈现出迅速升高、迅速降低的趋势;而AA-3则出现迅速升高、缓慢降低的趋势。在酿酒酵母发酵中,葡萄糖首先通过EMP途径代谢,为菌体生长提供ATP,而乙醇则作为该途径的终产物被大量分泌到发酵液中,作为一种未完全氧化的碳源储备;当葡萄糖等碳源耗尽之后,在有氧条件下,乙醇可以作为第二碳源被重新利用[21]。AA-1菌株和野生型菌株在24 h 生产了约7 g/L乙醇,随后便开始降低,AA-1的乙醇产量降低幅度更大;相比之下,AA-1的己二酸产量从24 h的 1.12 mg/L大幅升高到 48 h的3.39 mg/L。由以上现象可以推测,部分乙醇可能通过有氧呼吸作用转化为了己二酸。而AA-3由于TCA循环的中断,几乎无法进行乙醇的重新利用,因此在24 h 达到最大产量7.87 g/L后减少缓慢。
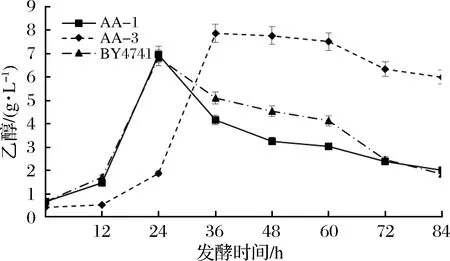
图7 菌株AA-1、AA-3和BY4741在YPD培养基发酵中乙醇的变化
为了对己二酸样品进行定性,将标准样品、野生型发酵样品及AA-1发酵样品进行液质联用定性检测,如图8所示。

特征离子:83、101、127、145;a-标准品;b-野生型发酵样品;c-AA-1发酵样品
己二酸在AA-1样品中检测到,特征离子为:83、101、127和145。值得一提的是,本研究仅仅在细胞破碎液中检测到了己二酸,而在发酵上清液中并未检测到。可能的原因是:1)酿酒酵母细胞壁缺乏己二酸的通道蛋白;2)己二酸总产量较低,无法形成足够的浓度梯度进行被动运输。
2.3 Tfu途径菌株的发酵碳源浓度控制
酿酒酵母具有高产乙醇的特性,这为酿酒酵母作为底盘生物合成高附加值代谢产物造成了一定的阻力:由于葡萄糖或其他碳源大量转化为乙醇,本应流向异源途径的代谢流切换到乙醇合成的方向,造成了资源的浪费,降低了目标产物的合成效率。在高糖浓度的情况下,即使在有氧条件下,酿酒酵母会将代谢由呼吸途径切换至发酵途径,大量生产乙醇,该现象被称为“Crabtree效应”[22];而若将酿酒酵母的乙醇合成途径相关基因如PDC1、ADH1基因敲除,会导致生长缓慢、代谢异常等现象[23]。从发酵工艺控制角度来看,乙醇的产生是Crabtree效应的具体表现,而促使酵母由呼吸切换至发酵途径的根本原因是碳源初始浓度。研究表明,随着碳源浓度的增长,Crabtree效应会愈加明显[24]。因此适当降低初始碳源的浓度是控制乙醇产量的一种方案。同时,Tfu途径始于乙酰辅酶A和琥珀酰辅酶A,两者均为有氧呼吸途径中产生的物质,因此本实验通过降低初始碳源浓度,降低Crabtree 效应,使酵母有氧呼吸作用增强,减少丙酮酸转换为乙醇的流量,使更多的碳源通过Tfu途径转化为目标产物己二酸。
本研究设置初始碳源质量浓度分别为5、10和 20 g/L,将AA-1菌株发酵48 h测定己二酸、乙醇、生物量等数据,并计算己二酸产率。随着初始碳源浓度的减少,乙醇产量显著下降(图9-a),但生物量(图9-b)和己二酸产量(图9-c)也随之下降。但己二酸产率则随着碳源浓度的降低而提高(图9-d)。

a-乙醇产量;b-生物量;c-己二酸产量;d-己二酸产率
在5 g/L 初始碳源浓度的条件下,己二酸产率比在20 g/L 初始碳源浓度的条件下提高了近1倍,说明初始碳源浓度的降低有助于Crabtree效应的减弱。
3 结论
本研究首次将T.fuscaB6 菌株中己二酸逆降解途径导入酿酒酵母体系,并实现了己二酸在酿酒酵母宿主中的异源合成。挑选了酿酒酵母内源组成型启动子、终止子,与6个外源基因进行搭配,成功构建了3个重组质粒并转化宿主细胞,实现了己二酸的合成。敲除TCA循环关键酶基因LSC1并未使己二酸产量获得提升。通过初始碳源浓度的控制,初步探究了己二酸产率的变化以及对副产物乙醇的控制。AA-1菌株在YPD培养基中发酵,得到了3.39 mg/L己二酸的产量,是同一宿主中报道的最高值。然而,目前该菌株合成己二酸的效率仍然较低。虽然在大肠杆菌中,利用该途径可以获得较高的己二酸产量,但在酿酒酵母宿主体系中,菌株合成己二酸的效率仍然较低。可能的原因如下:1)酵母细胞中途径基因的拷贝数低,导致途径酶表达量较低;2)酵母细胞中启动子强度较大肠杆菌相比偏低,导致途径酶表达量较低;3)碳源大量地流入乙醇及其他副产物的合成途径,造成己二酸的产量偏低;4)酵母特有的Crabtree效应导致呼吸向发酵代谢的切换,造成了碳源利用效率偏低;5)酵母属于真核生物,存在线粒体等亚细胞结构,当中进行的TCA循环所产生的己二酸前体代谢物——琥珀酰-CoA可能受到线粒体膜的空间分隔,不容易接触到胞质中的Tfu途径酶,造成途径催化效率低。在今后的研究中,我们将采取增加基因拷贝数、增强关键基因启动子强度、敲除或抑制竞争途径及发酵工艺优化等策略进一步提高己二酸的生产能力。
猜你喜欢
环境工程技术学报(2022年3期)2022-06-05 07:20:20
酿酒科技(2021年8期)2021-12-06 15:28:22
昆钢科技(2021年6期)2021-03-09 06:10:20
军事文摘·科学少年(2021年1期)2021-02-04 08:03:45
化工管理(2017年24期)2017-03-06 06:38:40
化工管理(2017年5期)2017-03-05 08:28:57
中国塑料(2016年1期)2016-05-17 06:13:12
故事作文·低年级(2016年7期)2016-05-14 09:39:46
电源技术(2016年9期)2016-02-27 09:05:25
中国塑料(2014年3期)2014-10-27 08:26:46
