
PCR-DGGE技术应用于南疆维吾尔族成人龋病、牙周炎口腔微生物多样性的研究
2020-04-30 04:18代海涛仵楠徐江唐小雪余芯乐李艳崔灵欣
石河子大学学报(自然科学版) 2020年2期
代海涛,仵楠,徐江,唐小雪,余芯乐,李艳,,崔灵欣
(1 石河子大学医学院第一附属医院,新疆 石河子,832008;2 石河子大学医学院,新疆 石河子,832008)
龋病与牙周炎的临床表现及疾病进程迥异,但都可以导致牙齿缺失[1]。长期以来这两种疾病常被独立研究[2],普遍认为龋易感者容易发生牙周炎,但事实并非如此。研究发现很多牙周状况较好的人确是龋敏感人群,而很多重度牙周炎患者,龋齿的患病率却很低,龋病和牙周炎的发生之间好似存在着相互抗衡的关系[3]。龋病与牙周炎均有明显的地区差异性和民族差异性[4],这也受到很多研究者的关注。新疆维吾尔民族与汉族在外部身材相貌,内部体格素质上有很大区别。南疆地区第三师51团固定人口5万余人,98.5%以上为维吾尔族,是维吾尔民族聚居区,生活饮食习惯遵从伊斯兰教风俗,生活区域较稳定,生活方式较单一,口腔保健意识及口腔医疗干预几乎为零。故为本课题组研究维吾尔族口腔微生物多样性提供了特色又稳定的研究背景。有研究认为维吾尔族人牙周病、龋病发生率均较高[5]。而本课题组前期研究发现,南疆维吾尔族人群患龋率高而牙周炎的患病率较低,且两者存在相互拮抗的现象,目前的研究很少涉及两者的发生环境与之相关性[6]。垂直凝胶电泳技术(Polymerase Chain Reaction-Denaturing Gradient Gel Electrophoresis,PCR-DGGE)在环境微生物多样性的研究中被广泛采用,具有操作方便快捷,可以批量操作、重复验证、价格适宜等优点。本研究从符合纳入标准的调查人群中筛查出高龋无牙周炎人群与无龋重度牙周炎组人群,通过PCR-DGGE技术分析该民族两组人群口腔微生物种类特点,从微生物角度了解这2种疾病有何关联,分析这2种疾病的发生机制,为预防及治未病提供新途径。
1 资料与方法
1.1 研究对象
针对南疆第三师51团采取随机抽样原则抽取309户居民,以家庭成员中35~50岁成人为研究对象,按照以下口腔检查方法分组。纳入标准:(1)南疆三代及以上常驻居民,无长期异地居住史;(2)无系统性疾病、慢性疾病以及先天性疾病;(3)口腔除龋病和牙周炎以外无其他疾病,牙齿结构及发育无异常;(4)口腔中无大面积修复体;(5)最近1个月内无抗生素及其它药物使用史;(6)非妊娠期或哺乳期。
1.2 口腔检查方法
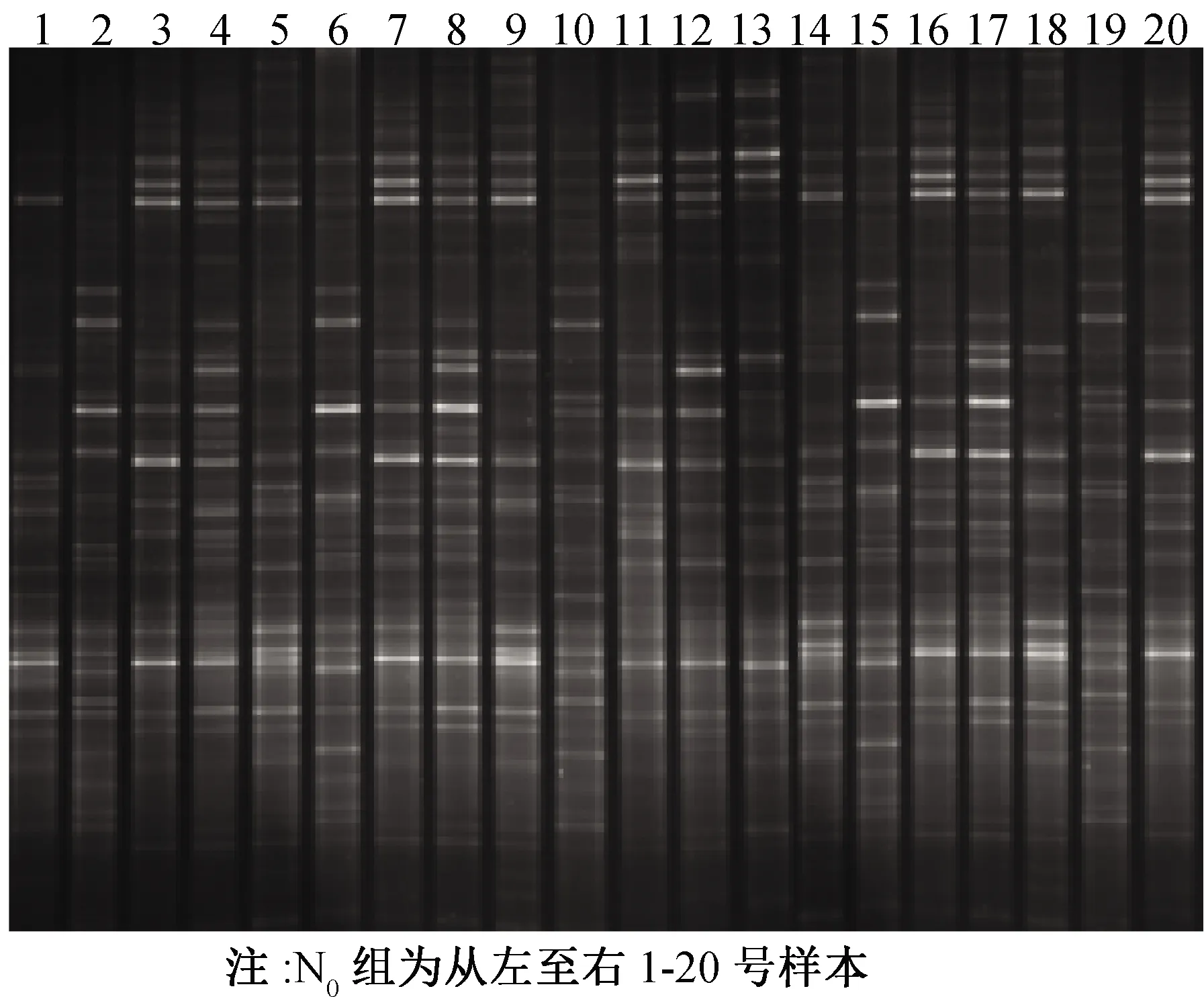
参考《第四次全国口腔健康流行病学调查方案》,3名口腔医师(标准一致性检验kappa>0.90)用牙周探诊及双头探针行牙周炎及龋病检查,记录患者龋失补牙数(decay missing fill,DMFT)。根据龋敏感程度[7],分为无龋组(DMFT=0、低龋组(0 研究对象晨起10∶00采集样本,之前用PBS液漱口30 s。用灭菌5 mL EP管收集研究对象自然排出的唾液2 mL,-4 ℃标记转运保存,-20 ℃冻存备用。 从健康组中抽取20例样本(N0组),高龋无牙周炎组中抽取10例样本(N1组),无龋重度牙周炎组中抽取10例样本(N2组)。各取3 μL进行琼脂糖凝胶电泳检测。16S V3区扩增引物357S-GCCGCCCGCCGCGCGCGGCGGGCGGGGCGGGGGCAC- GGGGGGCCTACGGGAGGCAGCAG;518R:ATTACCGCGGCTGCTGG[10],PCR反应体系(40 μL):2×Mix20 μL,上游引物1.0 μL,下游引物1.0 μL,DNA 模板2.0 μL,补ddH2O至40 μL。循环条件:94 ℃热启动2 min;94℃30 s,55 ℃退火30 s,72℃30 s,25个循环;72℃5 min。10 ℃保存[10]。Recondition PCR(5个循环)。1.50%琼脂糖,1×Tris-硼酸电泳缓冲液,电泳(120 V)30 min,核酸电泳染色。紫外线凝胶扫描成像,记录结果。 1.5.1 变性胶的制备 变性剂浓度52%~65%((100%变性浓度为:7 M Urea,40% Formamide),Bio-Rad Model475 Gradient Delivery System 灌胶系统以从顶部注入凝胶的填充方式进行梯度灌胶。 1.5.2 电泳及染色 凝固胶加样后,电压70 V,水浴温度60 ℃,电泳时间15.5 h。Biolinker DNA Red 染色液染色 40 min。 1.5.3 DGGE胶条带分析 染好的胶通过Bio-Rad 凝胶成像仪进行拍照,采用Quantity One分析软件(Bio-Rad),统计条带数,选用多维定标分析,非加权平均数(UPGMA,unweighted-pair group method arithmeticmean)对DGGE图谱进行聚类分析。 将DGGE胶上回收的条带胶4 ℃保存12 h,取3 μL为模板再次扩增,PCR扩增产物克隆测序。通过 NCBI- BLAST软件对所得序列进行分析,获得细菌的鉴定结果。 数据采用SPSS 19.0进行分析。采用χ2检验对纳入人群患龋率、慢性牙周炎患病率流行病学统计,采用方差分析及多重检验比较N0组、N1组、N2组PCR-DGGE条带数差异,P<0.05 表示差异有统计学意义。 本调查655名维吾尔族成年人群符合纳入标准的人为635人(无牙颌20例除外)(表1)。南疆第三师51团成年人龋病和牙周健康基本状况龋为5.12±2.07,患龋率为 92.28%;慢性牙周炎患病率为38.11%。高龋无牙周炎组与无龋重度牙周炎组流行病学分布有统计学差异(χ2=9.277,P=0.000*)。 表1 南疆第三师51团维吾尔族35~50岁成年人龋病、牙周炎流行病学分布 Reconditioning PCR扩增产物1.5%琼脂糖凝胶电泳的结果确认 N0组、N1组、N2组各样本的目的片段在250 bp,浓度适中(图1、图2)。 注:MarkerDL2000,其中亮带为20 ng/μL,其余为10 ng/μL。1-10为N1组样本,11-20为N2组样本图1 N1组、N2组16S V3区PCR检测电泳图 注:MarkerDL2000,其中亮带为20 ng/μL,其余为10 ng/μL。1-20为健康组样本图2 N0组16S V3区PCR检测电泳图 PCR-DGGE凝胶电泳条带清晰。N0组20例健康人群标本DGGE图谱条带计数总数为941条,平均数为46.90±2.53。N1组细菌条带技数总数为342条,平均为34.40±1.50。N2组细菌条带技数总数为294条,平均为29.90±3.14,方差分析多重检验比较发现各组间差异有统计学意义(P<0.000*)(图3、图4)。 图3 N0组PCR-DGGE检测电泳图 图4 N1组、N2组PCR-DGGE检测电泳 由Quantity One软件的非加权组平均法(UPGMA)所产生的系统发生树,其代表样品之间相似关系,通过该树可以直观的看到样品之间的相似性关系。 从图5中可以直观的看到N0组、N1组、N2组样本组内相似性较高(N1组样本1-10,N2组样本11-20,N0组样本为31-40),N0组与N1组、N2组之间的相似性较低,相似系数为0.05。 注:发育树中数值表示各样品之间的相似系数树枝末尾表示各样品编号图5 利用UPGMA方法构建的系统发生树 选取N1组样本1、2、3、4;N2组样本11、17、18、19;N0组样本31、33、35、36共12个样本经过切胶回收,克隆测序,BLAST检索条带序列和同源性比较后可以确定条带所代表的菌群,在GeneBank查询菌种。 N0组样本31、33、35、36中菌群成分相似,比例均衡,除bacteriodales所占比例较高以外,其余菌种较为平衡。N1组样本1、3、5、7样本以Streptococcaceae、Prevotellaceae、Lactobacillales、Veillonellaceae、Flavobacteriaceae为主,较N0组Veillonellacea、Flavobacteriacea、Prevotellaceae、Lactobacillales所占比例明显增高,而Streptococcaceae比例从1%~20%,分布不均匀。N2组样本11、17、18、19中Porphyromonadaceae、 Unclassfiled、 Leptotrichosis、Spirochaetaceae、Fusobacteriaceae为主,较N0组Porphyromonadaceae、 Unclassfiled、 Leptotrichosis、Spirochaetaceae、Fusobacteriaceae所占比例明显增高,但Lactobacillales比例明显下降(图6)。 牙周炎与龋病在世界各国发病率都较高,龋病被认为是世界第三大疾病,重度牙周炎更是给牙齿判了死刑。龋病与牙周炎都可以导致牙齿缺失,但两者发病特点和病损组织状态却大不相同。有研究认为细菌与宿主在外界环境变化时,口腔微环境平衡失调,微生物间此消彼长使得某些致病微生物占优势而致病[11-13],以往研究认为龋病多与变形链球菌、乳酸杆菌有关[14-15],牙周炎多与牙龈卟啉单胞菌等有关,长期以往对抗优势菌的治疗并未在防控龋齿及牙周炎上取得明显成效。而对于牙周炎与龋病的研究也常常被独立开来,两者之间是否存在协同相关、相互拮抗或者毫无关系等的流行病学和细菌学关系研究还鲜有报道。 微生物群落会受宿主的种族、生活环境、饮食习惯、生活方式的影响而变化。维吾尔族人群整体面貌偏向于欧罗巴人种,饮食习惯、生活方式独具特色。本研究选择生活环境较为封闭的南疆维吾尔族传统世居的35~50岁维吾尔成年人635名,经调查龋均为5.12±2.07,患龋率为 92.28%,明显高于全国平均水平82%[10];慢性牙周炎患病率为 38.11%,低于全国平均水平54%[16]。高龋无牙周炎组与无龋重度牙周炎组流行病学分布存在统计学差异(χ2=9.277,P=0.000)。本次研究发现,该人群普遍口腔卫生意识淡薄,能保障每日两次刷牙口腔保健的人微乎其微,牙菌斑、软垢及牙结石的检出率极高。从这一方面可以解释该因素是龋病患病率高,但牙周炎与龋病的发生不仅不是协同相加的关系,相反,牙周炎重的患者龋齿的患病率很低,而龋齿患病率高的患者牙周炎的患病率很低,两种疾病存在着负性关系,这与很多研究结果相反[17]。口腔中微生物数量众多,牙周炎患者与龋病患者口腔中微生物群构成是否有明显不同,两组菌群构成的趋向性和相互关系在目前国际国内还没有深入的研究。 Reconditioning PCR是指稀释原PCR产物至0.1倍,依照PCR原本反应体系,再次重复进行PCR扩增,与原PCR不同之处在于此次扩增仅需5个循环。在PCR反应中产生的异源双链对PCR-DGGE图谱的观察和分析会有很大干扰,而通过Reconditioning PCR可以有效清除异源双链。PCR-DGGE分析方法是一种较实用的现代分子生物学方法。应用PCR-DGGE技术,可以在一条泳道上分离出序列不同明暗强度不一的DNA条带,因此可以直观观察到微生物的丰富性,以及观察条带的明暗度反应微生物数量占比。v3区、v1-v3区、v6-v8区[18-19]在16Sr DNA中属于保守区域片段,多种微生物菌群的研究均采用。通过细菌16SrDNA可变区PCR,扩增产物通过DGGE可以显示出不同的条带,所以可以用 PCR-DGGE的方法进行区别。 本课题通过扩增16SrDNA V3可变区,应用PCR-DGGE技术,将健康组、高龋无牙周炎组与无龋重度牙周炎组3组人群唾液DNA中分离出多条电泳条带,且条带清晰明亮。健康组条带数明显多于其他两组,说明健康组口腔中微生物含量更为丰富,而其他两组口腔群落结构相对单一,这种结果可能与口内菌群平衡被打破时,有些细菌被替换导致缺失引起。关于健康组口腔微生物的研究比较广泛,而高龋无牙周炎组与无龋重度牙周炎组条带数据统计也存在明显差异,且这两组疾病人群口腔菌群的相互关系研究还很少见,故而本研究仅对两疾病组人群进行微生物分析。 生态学中许多物种或环境因子常常是是离散非连续性的,但许多统计学模型和计算方法往往是针对连续线性的模型。为了让非对称的物种数据可以用这些线性的统计模型,需要将数据进行转化。聚类分析也是需要先进行转化的。UPGMA可以用于分析分类问题,属于平均聚合聚类,是微生物多样性分析中常用的聚类分析方法。UPGMA最早是用来解决分类问题的。当用来重建系统发生树时其主要依据是聚到同一个数据集中的样本,将个体或者变量按相似程度划分类别,使得同一类中的元素之间的相似性比其他类的元素的相似性更强。目的在于使类间元素的同质性最大化和类与类间元素的异质性最大化。 本研究应用UPGMA方法计算遗传相似系数并做聚类图分析,结果显示健康组、高龋无牙周炎组、无龋重度牙周炎组人群组内相似系数较高,而健康组人群与其它两组人群组间相似系数仅为0.05,由此可以推断口腔疾病状态下微生物构成会发生重大变化。将分别属于同组且相似度较高的1、2、3、4、11、17、18、19、31、33、35、36样本PCR-DGGE产物回收、克隆、测序研究菌种之间的差异。结果发现健康组中除bacteriodales所占比例较高以外,其余菌种较为均衡,而bacteriodales正常寄居于人和动物的肠道、口腔、上呼吸道中。高龋无牙周炎样本中以Streptococcaceae、Prevotellaceae、Lactobacillales、Veillonellaceae、Flavobacteriaceae为主,较健康组Veillonellacea、Flavobacteriacea、Prevotellaceae、Lactobacillales所占比例明显增高,而Streptococcaceae并不是像以往研究那样占据绝对优势地位[14],其比例从1%~20%,分布不均匀。Streptococcaceae在无龋重度牙周炎组中也广泛存在,但比例偏低,也反向证明了Streptococcaceae与龋病呈正相关关系,但龋病的发生并不是由单一菌种导致,而是通过整个菌群结构改变所致。本研究发现Veillonellaceae、Flavobacteriaceae在高龋无牙周炎组中比例较高,在无龋重度牙周炎组中比例较低,推测Veillonellaceae、Flavobacteriaceae与龋病有正相关关系,这在以往的研究中少有提及。无龋重度牙周炎组样本以Porphyromonadaceae、 Unclassfiled、 Leptotrichosis、Spirochaetaceae、Fusobacteriaceae为主,较健康组Porphyromonadaceae、 Unclassfiled、 Leptotrichosis、Spirochaetaceae、Fusobacteriaceae所占比例明显增高,但Lactobacillales比例明显下降。Lactobacillales被认为是主要致龋菌,在高龋组中比例增高与很多研究一致[15],但在无龋牙周炎组中比例减少,也提示Lactobacillales可能是牙周组织健康的有益菌,这点并未在中外文献中查及。Porphyromonadaceae、Fusobacteriaceae一直以来都被认为是牙周炎的主要致病菌,与本次研究相符,但Unclassfiled在样本中比例较高,推测Unclassfiled在牙周炎中亦起到了重要作用,但在以往的文献中未能查及,这可能与研究方法有关。这些优势菌在唾液微生态环境起着什么作用有待进一步研究,菌种之间的相互制约或促进作用以及通过微生态的调节作用,能否逆转这种平衡从而达到防治龋病,牙周炎正是研究的热门和重要落脚点。 结合目前的文献以及本研究结果可以看出不同健康状态下口腔牙菌斑中的微生物群落结构及多样性具有较大的差异,菌群之间具有相当大的联系与区别,这也说明了口腔菌群与疾病有密切关系,菌群复杂性远超过优势菌群理论。PCR-DGGE技术可以批量研究多个样本的菌种,对大样本定性研究有绝对优势,但不能对各菌种定量,只能大概估计菌种构成比。后期课题组将针对样本进一步研究所测到的菌种数量的差异,微生物结构差异性进行研究,探讨细菌群落与疾病发展的关系,为口腔疾病防治提供微生物学身份信息。1.3 样本采集
1.4 DNA提取16S V3区扩增及检测
1.5 PCR-DGGE变性凝胶电泳图谱分析
1.6 回收条带、克隆测序及BLAST分析
1.7 统计学方法
2 结果与分析
2.1 纳入对象基本情况

2.2 16SV3区特征片段PCR结果


2.3 PCR-DGGE变性凝胶电泳图谱分析

2.4 健康组、高龋无牙周炎组与无龋重度牙周炎组微生物群落样品间的聚类分析

2.5 选取PCR-DGGE条带基因片段的测序分析
3 讨论
猜你喜欢
中国医学工程(2022年3期)2022-07-29
计算机应用与软件(2022年6期)2022-07-12
油气地质与采收率(2022年3期)2022-05-20
自然灾害学报(2022年2期)2022-05-10
中国药学药品知识仓库(2022年7期)2022-05-10
锦州医科大学报(2022年2期)2022-05-07
中国典型病例大全(2022年7期)2022-04-22
中国典型病例大全(2022年9期)2022-04-19
中国学校体育(2021年10期)2021-04-26
天津医科大学学报(2021年1期)2021-01-26
