
肺腺癌关键基因的生物信息学研究
2020-04-29 14:01刘冬琦窦涪琳杨晓东
中国实验诊断学 2020年4期
刘冬琦,窦涪琳,杨晓东*
(1.吉林大学中日联谊医院,吉林 长春130033;2.山东大学第二医院,山东 济南250033)
肺癌是世界上发病率和致死率最高的恶性肿瘤,肺腺癌(LUAD)目前已成为肺癌中最主要的病理类型,占所有肺癌患者的80%-85%[1]。尽管早期诊断和治疗方法取得了显著进展,但5年相对总生存率(OS)仍低于20%[2]。对于不能手术的癌症患者和手术患者,化疗仍然是最重要的辅助治疗,然而,药物不良反应及耐药性制约着化疗的最终效果。因此,迫切需要新的策略来补充传统的化疗[3]。在过去的几十年里,我们通过基因组学提高了对癌症分子特征的认知。晚期非小细胞肺癌的治疗策略已经从传统的基于组织病理学的化疗转变为基于致癌因素的个体化精确治疗[4]。虽然发现的生物标志物和治疗靶点对LUAD的诊断和治疗做出了巨大贡献,但由于LUAD的生物学复杂性和较差的预后,迫切需要更多的遗传信息,以提供精确医疗的参考[5]。为了探索与癌症相关的常见生物标志物和用于癌症治疗、诊断和预后的直接药物,近年来各国学者发布了诸多的癌症基因芯片和高通量测序数据[6-9]。同时,为了克服不同技术平台或小样本应用带来的局限性,生物信息学方法被广泛应用于癌症相关领域研究,发现了大量有价值的生物信息[10-12]。
应用GSE33532、GSE40791和GSE19188的基因芯片数据,利用生物信息学方法识别LUAD组织与正常组织之间的差异表达基因(DEGs)。此外,还建立了1354个hub基因和3个核心模块的蛋白质-蛋白质相互作用(PPI)网络。同时,通过Kaplan-Meier Plotter在线数据库发现NSCLC中有8个与OS相关的基因。此外,还对DEGs进行了富集分析。本研究旨在从新的角度识别与NSCLC发病和预后相关的关键基因。
1 材料方法
1.1 数据获取
我们在基因表达综合数据库(GEO,http://www.ncbi.nlm.nih.gov/GEO)筛选出GPL570数据集中包含肺腺癌样本及正常组织样本的基因表达谱数据(GSE33532、GSE40791、GSE19188、GSE31548、GSE43458),在R软件(3.5.2版,https://www.R-project.org/)环境下,使用affy/ affyPLM软件包生成各个基因表达谱数据的RNA降解图,筛选5’-3’逐渐升高的数据。最终3个表达谱数据纳入研究(GSE33532、GSE40791、GSE19188)。
所有数据都是基于Affymetrix人类基因组U133 Plus 2.0基因芯片,包含共计364例样本被纳入本项研究,其中包含179例肺腺癌样本和185例正常肺组织样本。
1.2 基因芯片数据分析
在R软件(3.5.2版,https://www.R-project.org/)环境下,使用affy软件包对数据预处理和识别、预处理和规范化。使用Limma包对每个GEO数据集的矩阵数据进行归一化和log2转换,每个基因芯片中的差异表达基因也由Limma 包进行筛选。|log2FC|≥2、调整P值<0.05为差异有统计学意义。
1.3 基因本体(GO)和京都基因与基因组百科全书(KEGG)富集分析
DAVID数据库(The Database for Annotation,Visualization and Integrated Discovery,https://david-d.ncifcrf.gov/home.jsp)是一个强大的基因功能分析工具,可对DEGs的GO进行注释及富集分析,去研究DEGs的生物功能,包括生物过程(BP)、分子功能(MF)和细胞成分(CC)。KEGG(https://www.kegg.jp/)可用于通路分析。P<0.05为存在显著性差异。
1.4 蛋白相互作用网络构建和模块分析
STRING数据库(STRING,https://string-db.org/)是一个探索PPI的在线工具。从STRING数据库中获得DEGs中的PPI信息,使用Cytoscape 3.7.1软件进一步构建。使用软件中的cytoHubba模块对所有DEGs进行筛选,并根据Degree进行排序,筛选出排名前十的基因作为核心基因(Hub genes),应用MCODE插件筛选出评分均>10的模块。并以各模块DEGs为基础进行KEGG富集分析。P<0.05存在显著性差异。
1.5 数据验证
基因表达谱交互分析(GEPIA)是一个在线网络工具,可以对癌症基因组图谱(TCGA)和基因型组织表达(GTEx)的肿瘤和正常数据进行基因表达分析,并将结果绘制成箱线图,我们可以在该网站调取Hub genes在LUAD组织中的表达情况来验证本研究数据的可靠性和重复性。
1.6 Hub genes的生存分析
生存曲线库 (K-M plotter,http://kmplot.com/)是一个在线数据库,基于GEO、TCGA数据库,分析肿瘤中某特定基因与死亡时间的关系,结果以95%置信区间和危险比来表示。本研究对Hub genes在肺腺癌中预后预测价值进行了分析。
2 结果
2.1 差异基因
在GEO数据库GPL570数据平台中选取的5个包含肺腺癌及正常肺组织基因芯片的数据集,并根据RNA降解情况(图1)选取了其中3个(GSE33532、GSE40791、GSE19188)共计364例样本被纳入本项研究,并对其中179例LUAD和185例正常肺组织进行了分析,共筛选出1354个DEGs(938个下调基因及416个上调基因),见图2。
2.2 差异基因的生物学功能注释
为了进一步了解差异基因的功能,应用DAVID数据库确定其GO分类及途径。结果表明,BP方面,下调的DEGs主要聚类于细胞黏附、生物附着、血管生成等,上调的DEGs主要聚类于M期、凋亡、细胞核分裂等。在分子功能方面,下调的DEGs主要聚类于碳水化合物结合、生长因子结合、结合方式等,上调的DEGs主要聚类于抗原结合、微管主动运动、金属内肽酶活性等。细胞成分分析结果显示,上调的DEGs主要聚类于纺缍体、染色体、浓缩染色体等,下调的DEGs主要聚类于细胞外、细胞膜,见图2。
2.3 DEGs的KEGG途径分析
如图3所示,上调的DEGs在细胞周期、p53信号通路、细胞外基质受体相互作用、卵母细胞减数分裂等通路中显著聚集。下调的DEGs主要富集于细胞黏附分子、血管平滑肌收缩、补体途径、黏着等。

图1 RNA降解图

图2 DEGs的GO及KEGG富集分析
2.4 PPI网络结构分析
在SRTING数据库的基础上,用Cytoscape软件构建出的蛋白相互作用网络,包含817个节点和5557条连线(图4),应用cytoHubba模块根据Degree筛选出前八个基因作为hubgenes,分别为CDK1、CDC20、CCNA2、CCNB1、BUB1、CCNB2、TOP2A及AURKB。应用MCODE插件共检测到3个评分>10的模块,对以上模块进行富集分析,其各自主要聚集途径这八个hub genes均在LUAD中高表达,且均在第一个模块内聚集(图5)。

图3 KEGG 富集分析
2.5 数据验证
应用GEPIA筛选了LUAD组织和正常组织之间的hub基因表达水平,图6反映了与正常组织相比,这8个基因在LUAD组织中的表达水平显著增高。

图4 PPI网络结构图,蓝色为上调基因,红色为下调基因

图5 三个核心模块及其KEGG富集情况
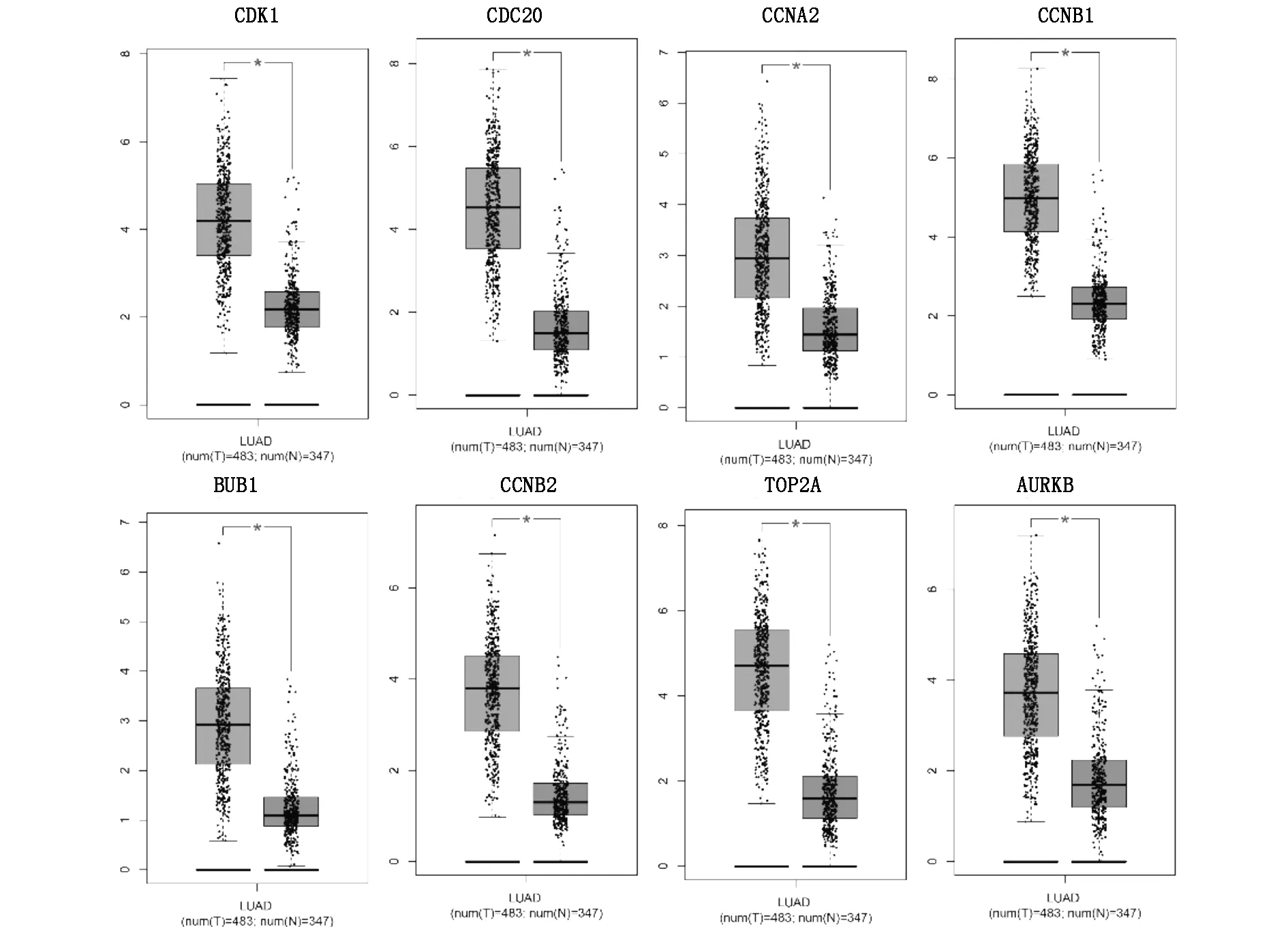
图6 hub genes在LUAD的表达情况
2.6 hub基因的预后价值
本研究采用K-M Plotter评价8个hub基因的预后预测价值。肺腺癌患者的总生存率是根据每个基因的高表达和低表达来计算的。腺癌患者中CDK1、CDC20、CCNA2、CCNB1、BUB1、CCNB2、TOP2A及AURKB高表达者OS更差,见图7。

图7 hub genes相关生存曲线
3 讨论
目前,肺腺癌已成为肺癌的最主要类型,然而,其发生和发展的潜在分子机制仍未充分阐明。本研究采用生物信息学方法预测肺癌的潜在治疗和预后评估靶点,并探讨肺癌可能的分子机制。本研究共筛选出1354个差异基因,其中938个下调基因,416个上调基因,通过构建PPI网络和富集分析,结合生存分析结果,共筛选出8个过表达关键基因CDK1、CDC20、CCNA2、CCNB1、BUB1、CCNB2、TOP2A及AURKB。在生物过程方面,下调的DEGs主要聚类于细胞黏附、生物附着、血管生成等,上调的DEGs主要聚类于M期、凋亡、细胞核分裂等,这与以前的认识一致,即细胞周期和细胞增殖调节因子的功能缺陷是肿瘤发生和发展的主要原因。根据Cavallaro等学者[13]的研究,细胞黏附分子表达的改变可以影响细胞的黏附功能、细胞的信号转导状态、细胞与环境的相互作用,并在肿瘤的进展、侵袭和转移中发挥重要作用。我们的结果提示,这些上调和下调的DEGs参与了这些BP,可能在NSCLC的进展中起重要作用。
本结果显示这8个过表达的hub genes 均与模块1相关,模块1在细胞周期途径中富集。表明这些基因均参与了细胞周期途径,并在癌症发展中发挥了重要的作用。
CDC20作为一种调节蛋白,在细胞周期的多个点上与其他几种蛋白质相互作用,它需要两个微管依赖的过程,后期的核运动和染色体分离。CDC20的高表达可预测肺癌患者甚至肺腺癌患者的预后不良[14]。CDC20的高表达与肺癌以外的许多癌症的预后不良相关,并且与肿瘤分级和分期相关[15]。
CCNB1作为参与有丝分裂的关键调控蛋白,在细胞周期G2/M转换中起着重要作用。Soria等人[16]的工作建立了CCNB1在非小细胞肺癌中的表达。结果表明,不同亚型的NSCLC不仅在生物学上存在差异,而且在CCNB1表达上也存在差异。在所检测的所有病理性分型中,CCNB1在鳞状细胞癌(SCC)中的过表达更为常见。这种过表达也会影响患者的生存时间,并可能成为SCC患者的不良预后标志物。细胞周期蛋白A2(CCNA2)是哺乳动物A型细胞周期蛋白家族中的一员。CCNA2控制细胞周期的G1/S和G2/M转换。在乳腺癌、肝癌、前列腺癌和肺癌等多种肿瘤中发现其蛋白表达升高,并可能成为预测生存率或早期复发的预后指标[17]。
Ⅱ型拓扑异构酶包含两种同工酶:TOP2A和TOP2B。在多种癌症中均检测到TOP2A的高表达,更重要的是TOP2A已成为公认的可用于临床中的癌症靶点。在乳腺癌、头颈部鳞状细胞癌和非小细胞肺癌,TOP2A在中、低分化肿瘤中的表达明显高于高分化肿瘤。TOP2A在NSCLC组织中的高表达水平与肿瘤的增殖、侵袭等恶性生物学行为密切相关,对TOP2A表达的干扰也可以抑制NSCLC细胞的增殖和侵袭[18]。
BUB1编码一种通过磷酸化有丝分裂启动子复合体的成员并激活纺锤体启动子而在有丝分裂中起无中心作用的碱/苏氨酸蛋白激酶。该基因的异常表达和突变与非整倍体和多种癌症有关。迄今为止,越来越多的证据表明,BUB1在各种癌症(包括胰腺癌和胃癌)中显著过表达与不良预后相关[19,20]。BUB1在不同类型癌症中不同作用的一个原因可能是不同的表达水平。然而,在本研究中,我们发现BUB1在LUAD中显著过表达,并与肿瘤不良预后相关,表明BUB1可能在LUAD的发生和发展中起作用。
AURKB是编码丝氨酸/苏氨酸激酶亚家族的一个成员,参与有丝分裂和减数分裂过程中染色体排列和分离的调节。已有研究证实,通过抑制AURKB,人肺癌细胞具有抗肿瘤和放射增敏作用[21]。
此外,我们研究的Kaplan-Meier绘图仪生存分析表明,这8个hub基因的mRNA表达水平与肺癌的临床预后显著相关。虽然这些可能暗示了它们在NSCLC进展中的作用,从而使它们成为NSCLC诊断和治疗的潜在靶点,但仍有必要对每个hub基因及其亚型的临床意义进行进一步的实验验证。
与以往的研究相比,本研究整合了来自多个数据集的相对较大样本量的基因芯片数据,并通过RNA降解图筛选出了质量较高的几组数据进行研究,结果可信度更高。虽然本研究通过生物信息分析,筛出了肺腺癌潜在的诊断和预后生物标志物,但结论仍有待于相应的实验证实。
猜你喜欢
昆明医科大学学报(2022年2期)2022-03-29
昆明医科大学学报(2022年1期)2022-02-28
皮肤性病诊疗学杂志(2021年5期)2021-11-27
现代临床医学(2021年6期)2021-11-20
三农资讯半月报(2021年1期)2021-01-27
浙江中西医结合杂志(2019年8期)2019-08-24
雷达学报(2017年6期)2017-03-26
CHINESE JOURNAL OF AERONAUTICS(2016年3期)2016-11-23
互联网天地(2016年1期)2016-05-04
现代计算机(2016年17期)2016-02-28
