
莪术醇的结构修饰以及抑制黑色素含量研究
2020-04-27 04:16陆雪莹黄烈军蹇军友苑春茂郝小江
天然产物研究与开发 2020年2期
陈 亮,陆雪莹,黄烈军,蹇军友,金 军,顾 玮,苑春茂,郝小江*
1贵州大学药学院,贵阳 550025;2贵州医科大学药用植物功效与利用国家重点实验室;3贵州省中国科学院天然产物化学重点实验室,贵阳 550014;4中国科学院干旱区植物资源化学重点实验室;5中国科学院新疆理化研究所,乌鲁木齐 830011
莪术为姜科植物莪术、郁金、广西莪术的干燥根茎,目前已大量人工栽培,资源充足,莪术醇为莪术油的主要活性成分,在莪术油中含量7%~8%,对莪术油药效的贡献最大[1]。文献报道莪术醇皮下注射对小鼠肉瘤S37、宫颈癌U14、艾氏腹水癌均有明显的抑制作用[2]。为了探索对黑色素的影响,我们选用黑色素瘤(B16)细胞,进行了莪术醇对细胞内黑色素含量的测定并以莪术醇为前体,对其化学结构进行修饰,合成了系列衍生物,以期发现新的抑制黑色素含量的化合物。
1 材料与方法
1.1 仪器与试剂
莪术油购自江西吉安市青原区友才天然香料油有限公司;实验所用试剂除特别说明外皆为市售分析纯试剂,且未经纯化直接使用;B16小鼠黑色素瘤细胞购自中国科学院细胞库,目录号TCM 2;核磁共振仪:INOVA-400和600型(Bruker公司,瑞士,TMS为内标);ESI-MS质谱仪:惠普LC-MSD型高效液相色谱-质谱联用仪。
1.2 莪术醇制备
将购得的2 kg莪术油经粗硅胶(40~80目)拌样后,进行柱层析色谱(石油醚∶乙酸乙酯=60∶1)的分离得到含莪术醇粗品,将所得到的粗品进行重结晶(乙醇),得到185 g白色晶体,易溶于氯仿。
1.3 莪术醇衍生物的制备
莪术醇衍生物的设计合成路线见图1。

图1 莪术醇衍生物的合成路线Fig.1 Synthetic route of curcumol derivatives注:试剂及反应条件(a)HBr,acetone,60 ℃(b)O3,Me2S,-78 ℃;(c)(BMIM)NTf2,(CH2O)n,i-Pr2NH·TFA;(d)Chloroacetic anhydride,NaH,DCM;(e)BOC-L-Leucine,NaHCO3,CSF,DMF;(f)m-CPBA,DCM;(g)NaOH,isopropyl alcohol;(h)NIS,PPh3,DCM;(i)Diethylamine,K2CO3,THF,70 ℃/NH3·H2O,40 ℃;(j)Carboxylic acid,DMAP,DCC,DCM;(k)4-Nitrobenzoyl chloride,NaH,KI,DMF。
1.3.1 2,7-二异丙基-1,4-二甲基-2,3-二氢环戊烷并环庚三烯-6(1H)-酮(1)的制备
在50 mL反应瓶中加入30 mL丙酮,再加入5 mL 40%的氢溴酸溶液,在此体系中加入无水硫酸钠干燥三小时,再加入五氧化二磷深度除水,过滤,得滤液[3]。将100 mg莪术醇溶于此滤液中,加热回流,TLC(5%的硫酸乙醇显色)跟踪至反应完全,加入饱和碳酸氢钠溶液中和体系中过量的溴化氢,无水硫酸钠干燥后减压浓缩,经柱色谱(石油醚∶乙酸乙酯=10∶1)得35 mg白色固体(1)。
1.3.2 (3S,3aS,5S,6R,8aR)-3-甲基-5-异丙基-7-甲烯基-8H-3a,6-环氧莪术-6-醇-8-酮(2)的制备
在50 mL反应瓶中加入20 mL二氯甲烷(DCM),加入100 mg莪术醇,置于低温反应器中,将温度设置为-78 ℃,在此温度下搅拌15 min[4]。随后,利用臭氧发生装置,向反应体系中通入臭氧,TLC(5%的硫酸乙醇显色)跟踪至反应完全,加入0.2 mL二甲硫醚,室温搅拌3 h后,依次用饱和亚硫酸氢钠,饱和食盐水洗,无水硫酸钠干燥后减压浓缩,经柱色谱(石油醚∶乙酸乙酯=10∶1)得48 mg无色透明液体(Z-1)。
在25 mL反应瓶中,依次加入2 mL 1-丁基-3-甲基咪唑二(三氟甲基磺酰)酰亚胺((BMIM)NTf2),中间体Z-1(19 mg,0.08 mmol),多聚甲醛(5 mg,0.16 mmol)及催化剂三氟乙酸二异丙基胺盐(i-Pr2NH·TFA)(18 mg,0.08 mmol),70 ℃下反应[5,6],TLC(5%的硫酸乙醇显色)跟踪至反应完全,依次用1 N的盐酸水溶液,1 N的氢氧化钠水溶液,饱和食盐水洗,无水硫酸钠干燥后减压浓缩,刮板,得17 mg白色固体(2)。
1.3.3 2-((3S,3aS,5S,6R,8aS)-3-甲基-5-异丙基-8-甲烯基八氢-6H-3a,6-环氧莪术-6-醇)-2-氧代乙基BOC-L亮氨酸(3)的制备
在50 mL反应瓶中,加入20 mL二氯甲烷 (DCM),加入莪术醇(300 mg,1.27 mmol)及氢化钠(NaH)(305 mg,12.7 mmol),室温搅拌30 min,随后加入氯乙酸酐(543 mg,3.2 mmol),室温搅拌[4],TLC(5%的硫酸乙醇显色)跟踪至反应完全,加水淬灭,依次用饱和酒石酸,饱和食盐水洗,无水硫酸钠干燥后减压浓缩,经柱色谱(石油醚∶乙酸乙酯=100∶1)得257 mg无色透明液体(Z-2)。
在25 mL反应瓶中,加入10 mL N,N-二甲基甲酰胺(DMF),再依次加入中间体Z-2(22 mg,0.071 mmol),BOC-L亮氨酸(17 mg,0.071 mmol),碳酸氢钠(NaHCO3)(6 mg,0.071 mmol)以及催化量的氟化铯(CSF),室温搅拌15 min,随后加热至70 ℃反应,TLC(5%的硫酸乙醇显色)跟踪至反应完全,加水淬灭,饱和食盐水洗,无水硫酸钠干燥后减压浓缩,经柱色谱(石油醚∶乙酸乙酯=30∶1)得28 mg无色透明液体(3)。
1.3.4 (3S,3aS,5S,6S,8aS)-3-甲基-5-异丙基-8-(N,N-二乙基-氨基甲基)-1,2,3,4,5,8a-六氢-6H-3a,6-环氧莪术-6-醇(4a)的制备
在100 mL反应瓶中,加入50 mL二氯甲烷(DCM),加入莪术醇(1 g,4.24 mmol)及间氯过氧苯甲酸(1.096 g,6.36 mmol),室温下搅拌[4],TLC(5%的硫酸乙醇显色)跟踪至反应完全,依次用饱和亚硫酸氢钠,饱和碳酸氢钠,饱和食盐水洗,无水硫酸钠干燥后减压浓缩,得粗品中间体(Z-3),将其溶于30 mL异丙醇中,加入氢氧化钠(512 mg,12.72 mmol),70 ℃下反应,TLC(5%的硫酸乙醇显色)跟踪至反应完全,依次用饱和酒石酸,饱和食盐水洗,无水硫酸钠干燥后减压浓缩,经柱色谱(石油醚∶乙酸乙酯=3∶1)得744 mg白色固体(Z-4)。
在50 mL反应瓶中,加入20 mL二氯甲烷(DCM),依次加入中间体Z-4(300 mg,1.2 mmol),N-碘代丁二酰亚胺(NIS)(405 mg,1.8 mmol)及三苯基膦(PPh3)(471mg,1.8mmol),室温下搅拌[7],TLC(5%的硫酸乙醇显色)跟踪至反应完全,饱和食盐水洗,无水硫酸钠干燥后减压浓缩,经柱色谱(石油醚∶乙酸乙酯=15∶1)得264 mg浅黄色液体(Z-5)。
在25 mL反应瓶中,加入10 mL 四氢呋喃(THF),依次加入中间体Z-5(100 mg,0.276 mmol),二乙胺(43 μL,0.414 mmol)及碳酸钾(K2CO3)(57 mg,0.414 mmol),70 ℃下反应[8],TLC(5%的硫酸乙醇显色)跟踪至反应完全,加水淬灭,饱和食盐水洗,无水硫酸钠干燥后减压浓缩,经柱色谱(石油醚∶丙酮=20∶1)得77 mg浅黄色透明液体(4a)。
1.3.5 (3S,3aS,5S,6S,8aS)-3-甲基-5-异丙基-8-氨甲基-1,2,3,4,5,8a-六氢-6H-3a,6-环氧莪术-6-醇(4b)的制备
在25 mL反应瓶中,加入中间体Z-5(132 mg,0.365 mmol)及14 mL氨水,40 ℃下反应[8],TLC(5%的硫酸乙醇显色)跟踪至反应完全,减压浓缩,经柱色谱(三氯甲烷∶甲醇=20∶1)得48 mg白色固体(4b)。
1.3.6 (3S,3aS,5S,6S,8aS)-3-甲基-8-羟甲基-5-异丙基-8-[(2-甲基-苯甲酰氧基)甲基]-1,2,3,4,5,8a-六氢-6H-3a,6-环氧莪术-6-醇(5a)的制备
在25 mL反应瓶中,加入10 mL二氯甲烷(DCM),再依次加入中间体Z-4(31 mg,0.4 mmol),邻甲基苯甲酸(20 mg,0.15 mmol),二环己基碳二亚胺(DCC)(76 mg,0.369 mmol)及催化量4-二甲氨基吡啶(DMAP),室温下搅拌[9],TLC(5%的硫酸乙醇显色)跟踪至反应完全,依次用饱和碳酸氢钠,饱和酒石酸,饱和食盐水洗,无水硫酸钠干燥后减压浓缩,经柱色谱(石油醚∶乙酸乙酯=10∶1)得32 mg无色透明液体(5a)。
1.3.7 (3S,3aS,5S,6S,8aS)-3-甲基-8-羟甲基-5-异丙基-8-[(3,5-二硝基-苯甲酰氧基)甲基]-1,2,3,4,5,8a-六氢-6H-3a,6-环氧莪术-6-醇(5b)的制备
在25 mL反应瓶中,加入15 mL二氯甲烷(DCM), 再依次加入中间体Z-4(103 mg,0.41 mmol),3,5-二硝基苯甲酸(104 mg,0.49 mmol),二环己基碳二亚胺(DCC)(253 mg,1.23 mmol)及催化量4-二甲氨基吡啶(DMAP),室温下搅拌[9],TLC(5%的硫酸乙醇显色)跟踪至反应完全,依次用饱和碳酸氢钠,饱和酒石酸,饱和食盐水洗,无水硫酸钠干燥后减压浓缩,经柱色谱(石油醚∶乙酸乙酯=10∶1)得106 mg白色固体(5b)。
1.3.8 (3S,3aS,5S,6S,8aS)-3-甲基-8-羟甲基-5-异丙基-1,2,3,4,5,8a-六氢-6H-3a,6-环氧莪术-6-醇对硝基苯甲酸二酯(6)的制备
在50 mL反应瓶中,加入15 mLN,N-二甲基甲酰胺(DMF),依次加入中间体Z-4(158 mg,0.627 mmol),氢化钠(NaH)(150 mg,6.27 mmol)及催化量碘化钾(KI),室温搅拌15 min,随后加入对硝基苯甲酰氯(348 mg,1.88 mmol),室温下搅拌[4,9],TLC(5%的硫酸乙醇显色)跟踪至反应完全,加水淬灭,依次用饱和酒石酸,饱和食盐水洗,无水硫酸钠干燥后减压浓缩,经柱色谱(石油醚∶乙酸乙酯=10∶1)得137 mg白色固体(6)。
1.4 莪术醇衍生物抑制黑色素含量的测定
所合成的莪术醇衍生物1、2、3、4a、4b、5a、5b、6,采用NaOH裂解法测定细胞内的黑色素含量[10]。将处于对数生长期的B16细胞消化,接种于6孔板中,浓度为2×105个/孔,并置于37 ℃、5% CO2培养箱中培养过夜。待细胞完全贴壁后用药,并继续培养72 h。弃去上清液,PBS洗涤2次,使用细胞刮收集细胞后每孔加入细胞裂解液200 μL。于4 ℃充分裂解40 min后,12 000 rpm离心20 min。转移上清至EP管测定蛋白浓度。沉淀物中加入190 μL 1 mol/L的NaOH(含10% DMSO)裂解液,并于80 ℃水浴1 h充分溶解后,在405 nm处测定吸光值,最终的黑色素含量使用相对蛋白浓度归一化处理。计算公式为黑色素相对含量=(OD405样品/蛋白浓度)/(OD405空白/蛋白浓度)×100%,每个样品重复3次进行测定。
2 结果与讨论
2.1 莪术醇的结果表征
1H NMR(400 MHz,CDCl3)δ:4.87(s,2H,H-9),2.55(m,2H,H-7),2.14(m,2H,H-4),1.00(d,J=4.0 Hz,3H,H-12),0.90(d,J=4 Hz,3H,H-11),0.86(d,J=4 Hz,3H,H-13);13C NMR(100 MHz,CDCl3)δ:144.8(C-8),113.0(C-9),104.7(C-6),88.2(C-3a),56.4(C-5),54.6(C-8a),39.5(C-3),38.9(C-7),34.8(C-4),31.0(C-2),28.8(C-10),28.3(C-1),23.2(C-11),21.6(C-12),12.5(C-13);HR-ESI-MS:m/z259.1667 [M+Na]+(calcd for C15H24O2Na,259.1669)。与文献[11-13]中莪术醇的核磁数据比较,确定所得晶体化合物为莪术醇。
2.2 莪术醇衍生物的结果表征
化合物1白色固体,收率33%。1H NMR(400 MHz,CDCl3)δ:7.08(s,1H,H-6),7.00(s,1H,H-9),3.46(hept,J=6.8 Hz,1H,H-11),3.07(p,J=7.0 Hz,1H,H-4),2.81 (dd,J=16.7,7.7 Hz,1H,H-2),2.54(dd,J=16.7,11.5 Hz,1H,H-2),2.22(s,3H,H-14),1.92(m,1H,H-3),1.73(m,1H,H-6),1.17(d,J=3.1 Hz,3H,H-15),1.16(d,J=3.1 Hz,3H,H-12 or H-13),1.04(d,J=7.1 Hz,3H,H-12 or H-13),0.99 (d,J=2.0 Hz,3H,H-17 or H-18),0.98(d,J=2.2 Hz,3H,H-17 or H-18);13C NMR (100 MHz,CDCl3)δ:185.3(C-8),158.6(C-7),151.8(C-5),144.9(C-1),144.8 (C-10),138.8(C-9),130.7(C-6),48.7(C-3),46.7(C-4),38.6(C-2),29.7(C-11),28.3(C-16),25.0(C-14),22.6(C-12),22.4(C-13),21.7(C-17),21.6 (C-18),14.2(C-15);HR-ESI-MS:m/z281.187 3 [M+Na]+(calcd for C18H26ONa,281.187 6)。
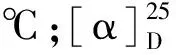
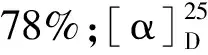
化合物4a浅黄色透明液体,收率90%。1H NMR(600 MHz,CDCl3)δ:5.76(s,1H,H-7),3.10(d,J=13.1 Hz,1H,H-9),2.77(d,J=13.5 Hz,1H,H-9),2.61(s,2H,H-1′),2.40(s,2H,H-1′′),1.02(d,J=6.7 Hz,12H,H-11,H-12,H-2′ and H-2′′),0.90(d,J=6.6 Hz,3H,H-13);13C NMR(150 MHz,CDCl3)δ:143.1(C-8),126.0(C-7),103.4(C-6),87.0(C-3a),59.5(C-9),56.8(C-5),50.3(C-8a),47.0(C-1′ and C-1′′),40.3(C-3),36.7(C-4),31.2(C-2),31.0(C-10),27.4(C-1),22.7(C-11),21.4(C-12),11.8(C-13),11.6(C-2′ and C-2′′);HR-ESI-MS:m/z308.258 4 [M+H]+(calcd for C19H34NO2,308.258 4)。
化合物4b白色固体,收率53%。1H NMR(600 MHz,CDCl3)δ:7.97(s,2H,N上氢),6.15 (s,1H,H-7),3.72(d,J=14.6 Hz,1H,H-9),3.63(d,J=15.5 Hz,1H,H-9),1.01(d,J=5.9 Hz,3H,H-12),0.99(d,J=6.5 Hz,3H,H-11),0.86(d,J=6.0 Hz,3H,H-13);13C NMR(150 MHz,CDCl3)δ:136.33(C-8),127.54(C-7),103.83(C-6),87.33(C-3a),58.62(C-5),50.53(C-8a),42.20(C-9),40.17(C-3),35.94(C-4),31.30(C-2),30.44(C-10),27.52(C-1),22.68(C-11),21.90(C-12),11.82(C-13);HR-ESI-MS:m/z252.195 4 [M+H]+(calcd for C15H26NO2,252.195 8)。
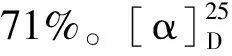
2.3 细胞内黑色素含量测定
表1 细胞内黑色素含量测定
经Grphpad prism软件分析,各组之间对比,均无显著性差异,其中与阴性对照组相比,化合物1、2、3、4a、4b、5a、5b、6以及莪术醇的P值分别为0.94、0.78、0.63、0.60、0.74、0.40、0.19、0.13、0.23。由含量测定结果首次发现,先导化合物莪术醇具有一定美白作用,8个衍生物中有2个衍生物的抑制黑色含量的作用超过莪术醇。中间体Z-4,9位羟基上接入3,5-二硝基苯甲酸(5b),成酯键后作用比莪术醇强;中间体Z-4,9位羟基和半缩酮羟基上都接入对硝基苯甲酰氯(6)成酯键后作用也比莪术醇强,而且也比化合物5b的作用强。而中间体Z-4,9位羟基上接入邻甲基苯甲酸(5a),成酯键后作用则明显降低,比莪术醇弱。由此可见Z-4中间体羟基上引入苯环上具有吸电子基团的苯甲酸成酯键后的抑制黑色含量的作用要优于引入苯环上具有给电子基团的苯甲酸。莪术醇在酸性条件下发生开环重排得到的衍生物1,针对半缩酮羟基的修饰,得到的衍生物3,莪术醇经过臭氧化,再经过mannich反应,得到具有α,β不饱和酮结构的衍生物2,以及引入含氮基团得到的衍生物4a和4b,其作用均比莪术醇弱。
3 结论
本文首次发现莪术醇具有抑制B16细胞中黑色素的活性,具有一定的美白作用。以莪术醇为前体,制备了8个系列衍生物,其中2、3、5a、5b、6为新化合物,经抑制黑色素含量的测定,其中衍生物5b和6的抑制黑色素含量的作用要优于莪术醇。经初步的构效关系分析可知,在中间体Z-4羟基上引入苯环上具有吸电子基团的苯甲酸成酯键后的抑制黑色素含量的作用要优于引入苯环上具有给电子基团的苯甲酸。研究结果对于莪术醇的结构修饰及其美白作用的深入研究具有一定的参考价值。
猜你喜欢
长春中医药大学学报(2022年6期)2022-11-23
盐科学与化工(2022年3期)2022-04-11
农技服务(2020年7期)2020-07-21
天津农业科学(2020年11期)2020-01-03
中国绿色画报(2018年4期)2018-08-09
绿色科技(2017年18期)2017-11-01
热带农业工程(2017年2期)2017-08-29
吉林农业(2017年5期)2017-05-13
江苏农业科学(2015年1期)2015-04-17
中国医学创新(2013年2期)2013-03-28
