
间二甲苯分子在不同外电场下结构和解离特性研究
2020-04-25 06:38:46向前进尹文怡刘玉柱张程元喆布玛丽亚阿布力米提
原子与分子物理学报 2020年1期
向前进, 尹文怡,2, 茆 锐, 刘玉柱,2, 张程元喆, 布玛丽亚·阿布力米提
(1.南京信息工程大学江苏省大气海洋光电探测重点实验室, 南京210044;2.江苏省大气环境与装备技术协同创新中心, 南京210044;3.常州工学院数理与化工学院, 常州 213032;4.新疆师范大学物理与电子工程学院, 乌鲁木齐830054)
1 引 言
随着工业化的发展,人类排放在大气中的挥发性有机物(VOCs)大量增加. VOCs挥发性强,来源极广. 光物理光化学反应,生物排放以及人为排放源均可引起VOCs浓度变化[ 1]. 其中,太阳光辐射下的光物理光化学反应促进了污染物质向二次污染物质的转变,对人类赖以生存的大气环境构成了严重的影响,也危害到了人类的健康. 研究表明,VOCs与肺癌、肺损伤、纤维化肺泡炎、肺功能异常等疾病有密切关系[ 2-7]. 开展对VOCs关键成分的研究,有利于保护大气环境,维护人类健康.
间二甲苯是VOCs的关键活性组分[ 8],研究如何通过降解间二甲苯以达到对VOCs排放的控制,减小其对大气环境与人类健康的影响是当今的热点问题. 关于间二甲苯的降解,代表性工作有:许锐伟等人以废水处理厂的污泥为菌源,通过筛选培育得到目的菌株,研究了在非离子型表面活性剂地强化作用下混合菌对于间二甲苯的降解[ 9];Korologos等人研究了不同紫外光照射下,TiO2催化剂对间二甲苯的催化氧化[ 10]. 但关于间二甲苯在外电场下的降解鲜有文献报道. 当分子置于外加电场中时,会发生一系列化学及物理变化,如吸收光谱改变,出现振动斯塔克效应等[ 11].
本文采用密度泛函的理论,在B3LYP/6-311G++基组水平上研究了不同外电场作用下间二甲苯分子的结构与解离特性. 为在外电场作用下,对间二甲苯分子进行降解提供重要的理论参考. 二甲苯体系都容易断裂一个甲基(CH3)发生解离,因此本文主要研究处于电场增加方向的甲基与苯环之间起连接作用的C-C键的降离.
2 计算方法
外电场作用下分子体系的哈密顿量H为:
H=H0+Hint
(1)
其中H0为无外电场时的哈密顿量,Hint为外电场与分子体系相互作用的哈密顿量. 在偶极近似的条件下,分子体系与外加电场的相互作用势能为:
Hint=-μF
(2)
其中,μ为分子的电偶极矩. 本文选取的电场区间为(-0.025 a.u. ~ 0.025 a.u.),且1 a.u. =5.14225×1011V/m.本文所涉及的计算,均在量子化学计算软件Gaussian09[ 12 ]中进行. 通过不同基组计算得到间二甲苯分子红外光谱,然后与实验值进行比较,选取与实验值吻合最好的B3LYP/6-311G++基组,并在该基组水平上计算了分子的几何构型,总能量,前线轨道能级,红外光谱以及解离势能面等.
3 结果与讨论
3.1 无外电场时的分子稳定构型,基组选择与矫正因子
理论计算表明间二甲苯分子为C1点群分子,本文采用密度泛函理论,在B3LYP方法下选用不同的基组对间二甲苯分子进行优化,得到了相应的红外光谱曲线. 对计算所得的红外光谱进行矫正,并与实验红外光谱[ 13]进行比较,选出最优基组.
3.1.1 分子稳定构型与基组选择
本文采用密度泛函理论,在B3LYP方法下选取了5种不同的基组对间二甲苯分子进行了优化计算,计算获得的红外光谱特征峰频率展示于表1中,并与实验值进行比较. 线性相关系数越接近1说明两者相关性越好,由表1可见,所选基组的相关系数R2都十分接近于1,说明选取B3LYP方法对间二甲苯进行理论计算具有极高的精确性与可行性. 通过对比,我们发现采用B3LYP/6-311G++基组计算得到的特征峰频率与实验结果的相关性最好,相关系数R2达到了0.99887. 因此本文中关于间二甲苯结构与解离特性的计算均在该基组下进行,计算得到的间二甲苯分子稳定构型见于图1. 本文沿间二甲苯分子两甲基中C原子连线方向施加不同强度的电场,即图1中箭头指示的方向. 采用B3LYP/6-311G++基组计算得到的红外光谱特征峰频率与实验值的线性拟合结果见于图2.

表1 实验和计算红外光谱的相应峰值频率及其相关系数
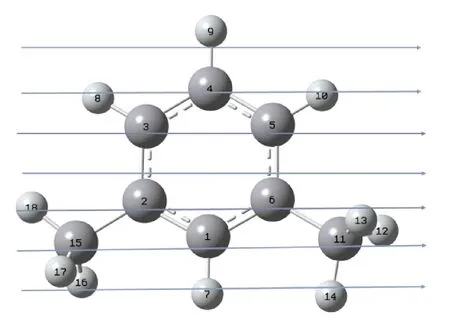
图1 间二甲苯分子稳定构型(箭头代表电场施加的方向)Fig. 1 The optimized geometry of ground state of m-xylene and the arrows represent the direction of the applied external electric field

图2 实验特征峰频率与计算特征峰频率的线性拟合结果Fig. 2 The linear fitting result of computational peak frequencies with experimental values
3.1.2 矫正因子与红外光谱
首先,通过下式[ 14]计算矫正因子:
(3)
其中C为频率矫正因子,wi为计算频率,νi为实测频率,Nf为选用的频率数. 将表1中B3LYP/6-311G++基组数据和实验数据代入(3)式,得到的频率矫正参数为0.964999073. 经矫正因子修正后的计算红外光谱与实验红外光谱展示于图3. 将修正后的红外光谱图与实验红外光谱对比,发现两者的峰值频率与特征谱线极其相似. 说明利用密度泛函B3LYP/6-311G++方法获得的红外光谱具有极高的准确性. 同时由于计算光谱具有较高分辨率,可以为实验红外光谱分析提供有效参考.

图3 实验红外光谱与矫正红外光谱Fig. 3 The experimental IR spectrum and corrected computational IR spectrum
3.2 外电场对分子总能量,键长,电偶极矩的影响
沿两甲基中C原子连线方向施加不同强度的电场(-0.025 a.u. ~ 0.025 a.u.),在B3LYP/6-311G++基组水平上对间二甲苯分子进行结构优化与计算,得到了不同外加电场下间二甲苯的分子总能量,键长和电偶极矩.
如图4所示,随着两甲基中C原子连线方向电场强度的增加,间二甲苯分子总能量先增大后降低. 且在无外电场时能量最高,表明此时越不易发生化学反应. 施加电场后,分子能量降低,同时可观察到,电场强度越大时,总能量变化的越快,表明外加电场对分子的总能量影响也越大.
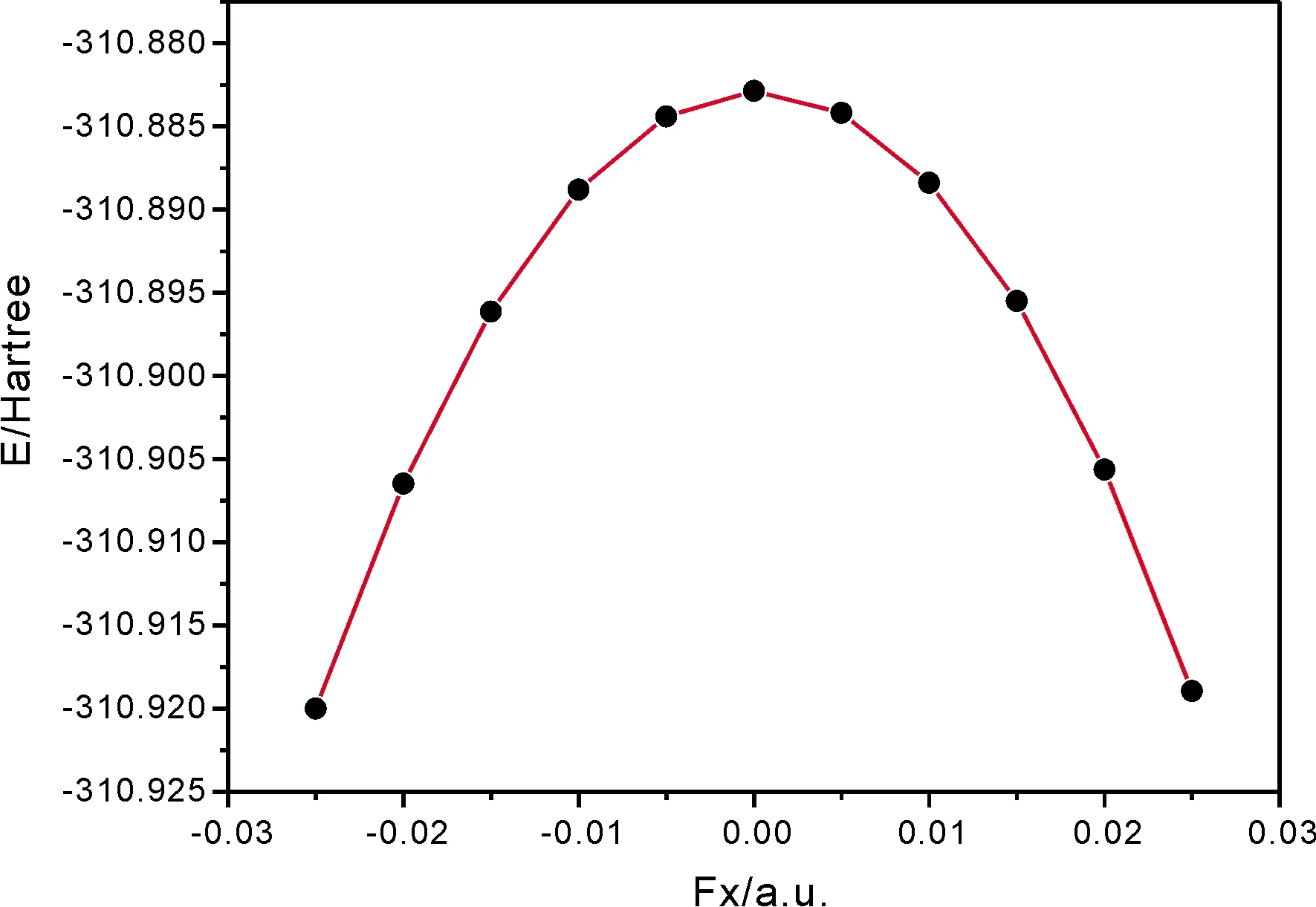
图4 间二甲苯分子总能量随外电场变化Fig.4 The variations of total energy of m-xylene under different external fields
由表2中数据可知,外加电场对间二甲苯键长影响情况不同,其变化趋势见图5. 随着两甲基中C原子连线方向正向电场强度增加,C2-C15键缩短,说明该化学键随着电场强度的增加变得越来越稳固. 而此时C6-C11键随着外加电场强度的增加逐渐伸长,说明该键越来越脆弱,越容易发生解离. 当施加负向电场时,变化情况刚好相反,此时C2-C15键越来越脆弱,越容易发生解离,而C6-C11键越来越稳固. 同时可以观察到,随着负向电场的增加,C1-C6键缓慢伸长,越来越容易发生接离,而施加正向电场时C1-C6键键长几乎没有变化,说明此时电场对它的影响很小.
图6展示的是外加电场对间二甲苯分子电偶极矩的影响. 随着两甲基中C原子连线方向电场的强度(-0.025 a.u. ~ 0.025 a.u.)增加,间二甲苯分子的电偶极矩先单调减小后单调增加,分子极性先变小后变大.
3.3 外电场对轨道能级分布的影响
在两甲基中C原子连线方向施加不同电场强度的电场,采用B3LYP/6-311G++基组对间二甲苯分子进行能量计算. 通过计算得到的间二甲苯分子在不同外电场作用下最低空轨道(LUMO)能量EL,最高占据轨道(HOMO)能量EH和能隙EG,如表3所示. 表中能隙按照下式计算得到
表2 不同外电场下间二甲苯的键长
Table 2 The bond lengths of m-xylene under different external fields
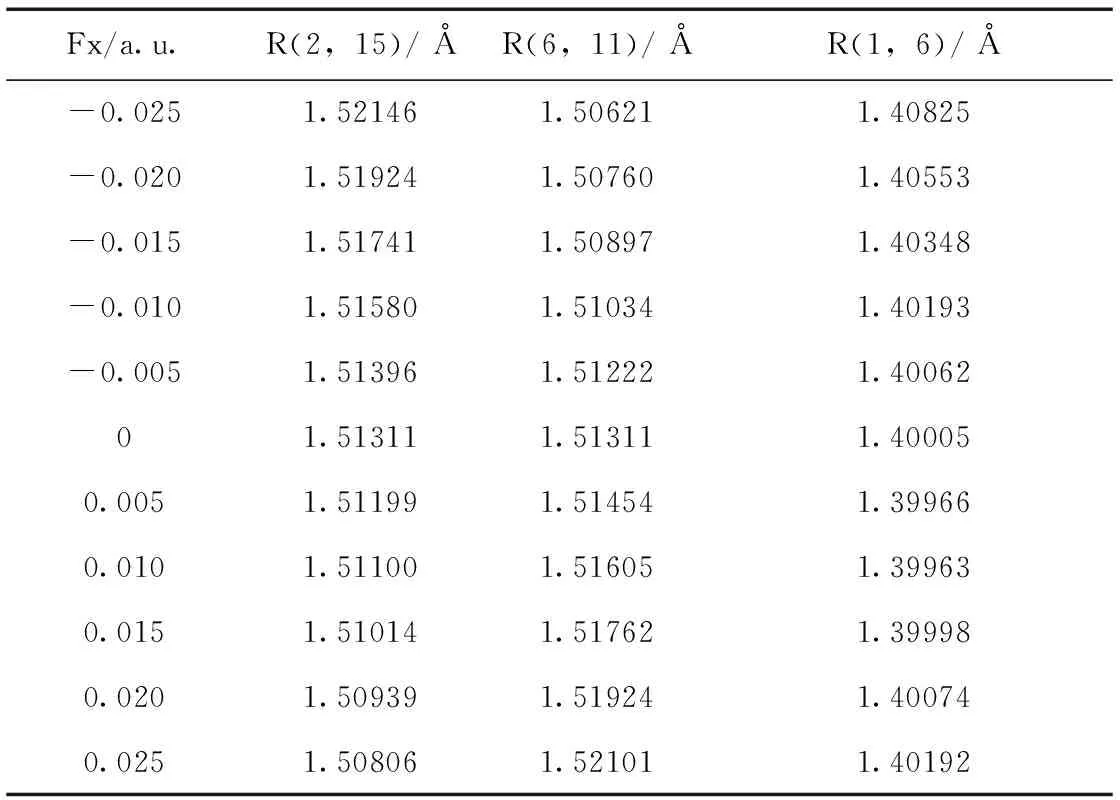
Fx/a.u.R(2,15)/ ÅR(6,11)/ ÅR(1,6)/ Å-0.0251.521461.506211.40825-0.0201.519241.507601.40553-0.0151.517411.508971.40348-0.0101.515801.510341.40193-0.0051.513961.512221.4006201.513111.513111.400050.0051.511991.514541.399660.0101.511001.516051.399630.0151.510141.517621.399980.0201.509391.519241.400740.0251.508061.521011.40192
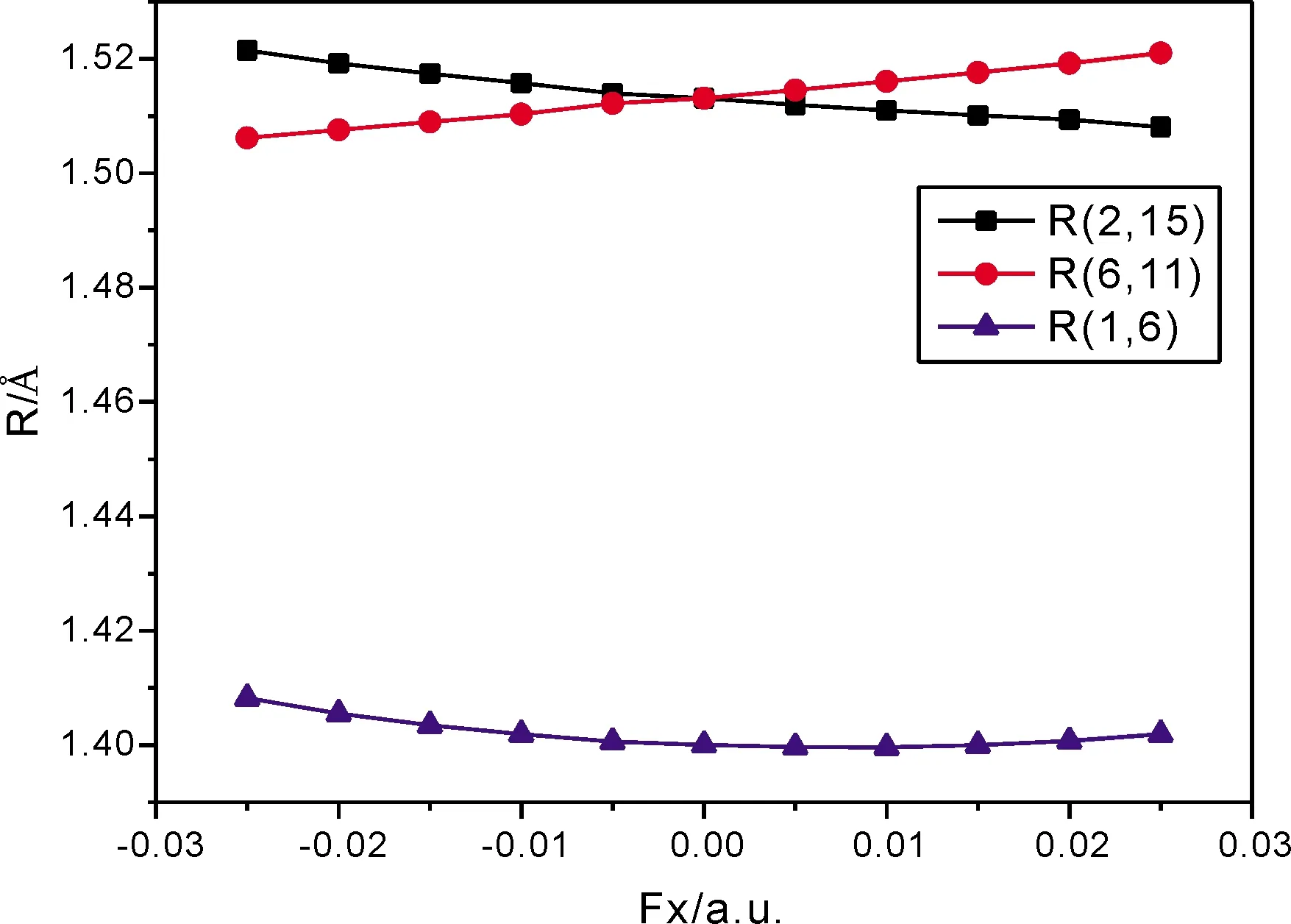
图5 间二甲苯分子键长随外电场的变化Fig. 5 The variations of the bond lengths of m-xylene under different external fields
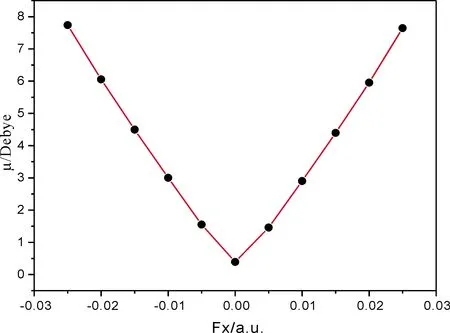
图6 间二甲苯分子电偶极矩随外电场变化情况Fig. 6 The variations of dipole moment of m-xylene under different external fields
表3 不同外电场下间二甲苯分子最低空轨道能量EL和最高占据轨道能量EH,能隙EG
Table 3 The LUMO energiesEL, HOMO energiesEH, energy gapsEGunder different external fields
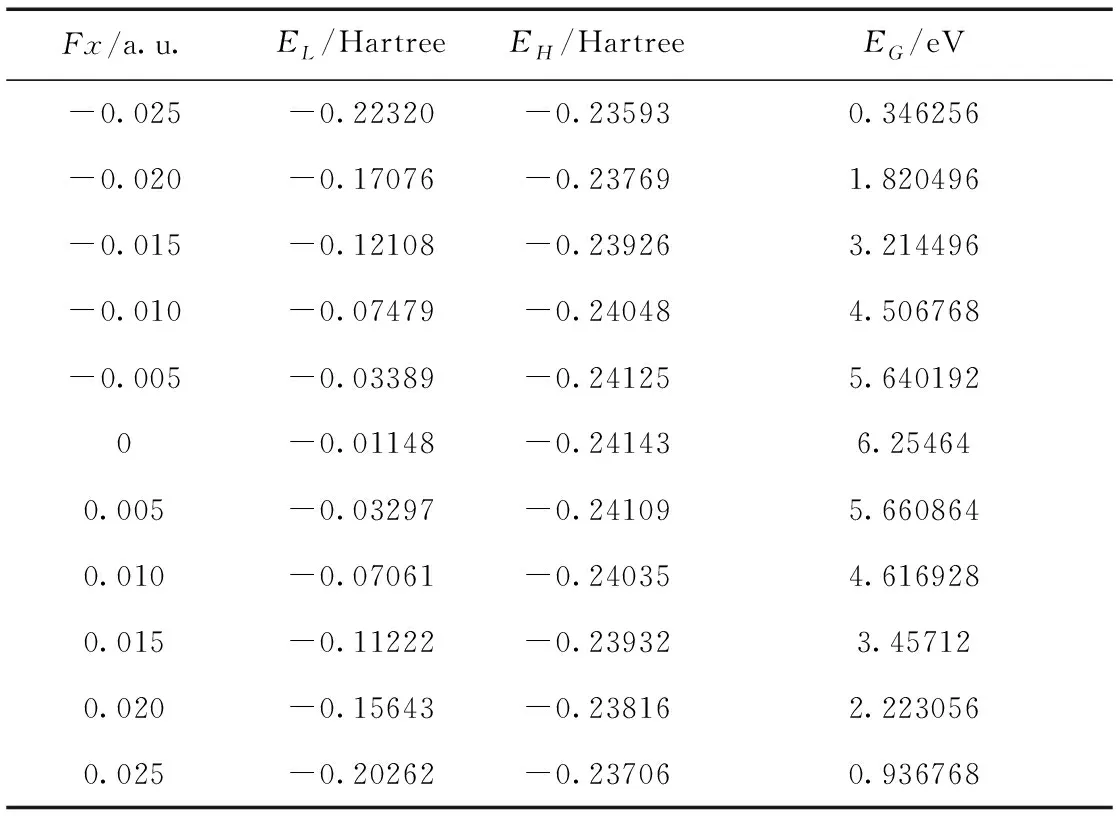
Fx/a.u.EL/HartreeEH/HartreeEG/eV-0.025-0.22320-0.235930.346256-0.020-0.17076-0.237691.820496-0.015-0.12108-0.239263.214496-0.010-0.07479-0.240484.506768-0.005-0.03389-0.241255.6401920-0.01148-0.241436.254640.005-0.03297-0.241095.6608640.010-0.07061-0.240354.6169280.015-0.11222-0.239323.457120.020-0.15643-0.238162.2230560.025-0.20262-0.237060.936768
EG=(EL-EH)× 27.2114 eV
(4)
分子最低空轨道能量和最高占据轨道能量代表着分子得失电子的能力.EL越小,分子越容易得到电子,EH越大,分子越容易失去电子. 由表3可知,随着两甲基中C原子连线方向外加电场强度的(-0.025 a.u. ~ 0.025 a.u.)增加,间二甲苯分子的最低空轨道能量先增大后减小,说明间二甲苯分子获得电子的能力先减弱后增强. 同时可在表3观察到最高占据轨道能量在外电场下没有发生明显变化,说明外电场对间二甲苯分子最高占据轨道能量的影响较弱. 其变化趋势见图7.
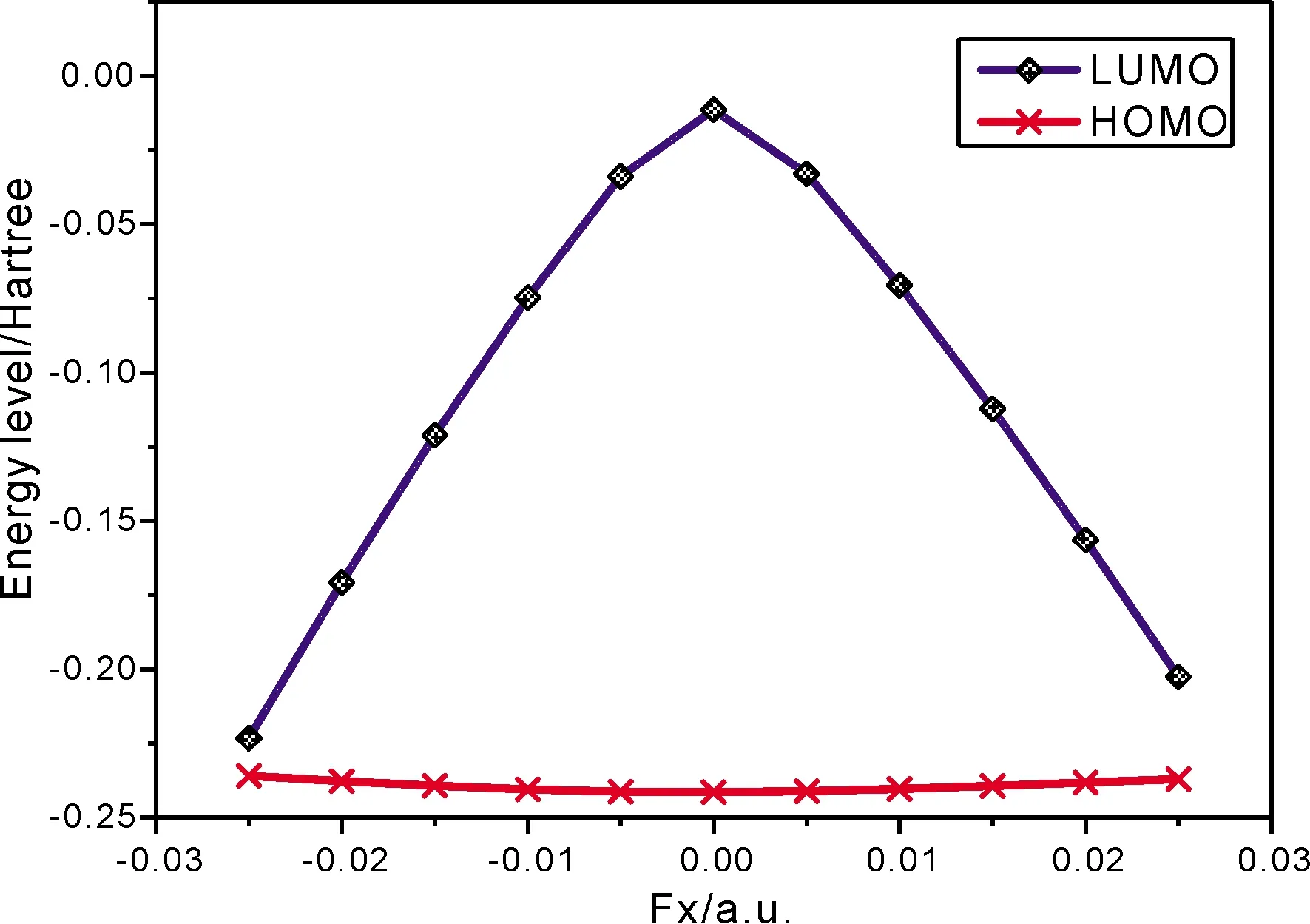
图7 不同外电场下间二甲苯分子EL与 EH的变化情况Fig. 7 The variations of EL and EH of m-xylene under different external fields
由图8可知,随着两甲基中C原子连线方向外加电场强度(-0.025 a.u. ~ 0.025 a.u.)的增加,间二甲苯分子能隙先增大后变小,且施加电场后,分子能隙均比无外电场时要小,说明施加电场后,间二甲苯分子越容易被激发到激发态而参与或发生化学反应.
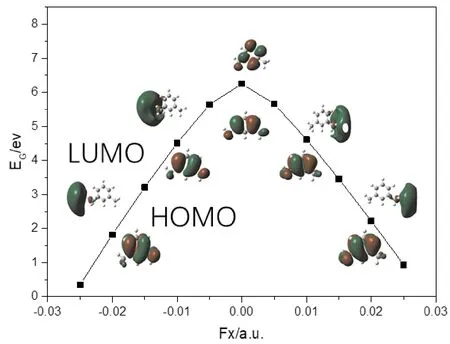
图8 间二甲苯分子能隙EG与轨道随外电场变化情况Fig.8 The variations of energy gap and orbit ofm-xylene under different external fields
图8还显示了电场强度分别为0,±0.01 a.u.和±0.02 a.u.时,间二甲苯分子的最低空轨道(LUMO)和最高占据轨道(HOMO). 当无外加电场时,电子云均匀分布在间二甲苯分子周围. 随着沿两甲基中C原子连线方向正向电场强度的增加,由于外部电场对电子云的吸引作用,间二甲苯分子的最低空轨道(LUMO)向电场增加的方向移动,而最高占据轨道(HOMO)恰好向相反方向移动,施加负向电场时,间二甲苯分子的最低空轨道与最高占据轨道变化规律亦同. 如图8所示,仍可直接观察到外电场对最低空轨道的影响远远大于最高占据轨道的影响.
3.4 外电场对红外光谱的影响
在B3LYP/6-311G++基组水平上对间二甲苯进行优化和红外光谱计算,获得了间二甲苯分子的红外光谱. 伸缩振动的红外强度与化学键发生断裂的难易程度密切相关. 在这里我们研究了受外电场影响很大的两种模式的伸缩振动. 分别为:频率为3008.48 cm-1的C11甲基团对称伸缩振动,频率为3156.58 cm-1的C3-C8与C5-C10键的伸缩振动.
在甲基中C原子连线方向上施加不同强度的电场(0 ~ 0.025 a.u.),得到的两种模式振动的频率变化情况如图9所示,随着电场强度(0 ~ 0.025 a.u.)增加,C3-C8与C5-C10键伸缩振动的频率明显减小,光谱呈现红移,同时振动强度亦随着电场强度的增加而增大, C11甲基团的对称伸缩振动频率也明显减小,光谱呈现红移,但振动强度先减小后快速增加,且在电场强度为0.005 a.u. 时振动强度最小.
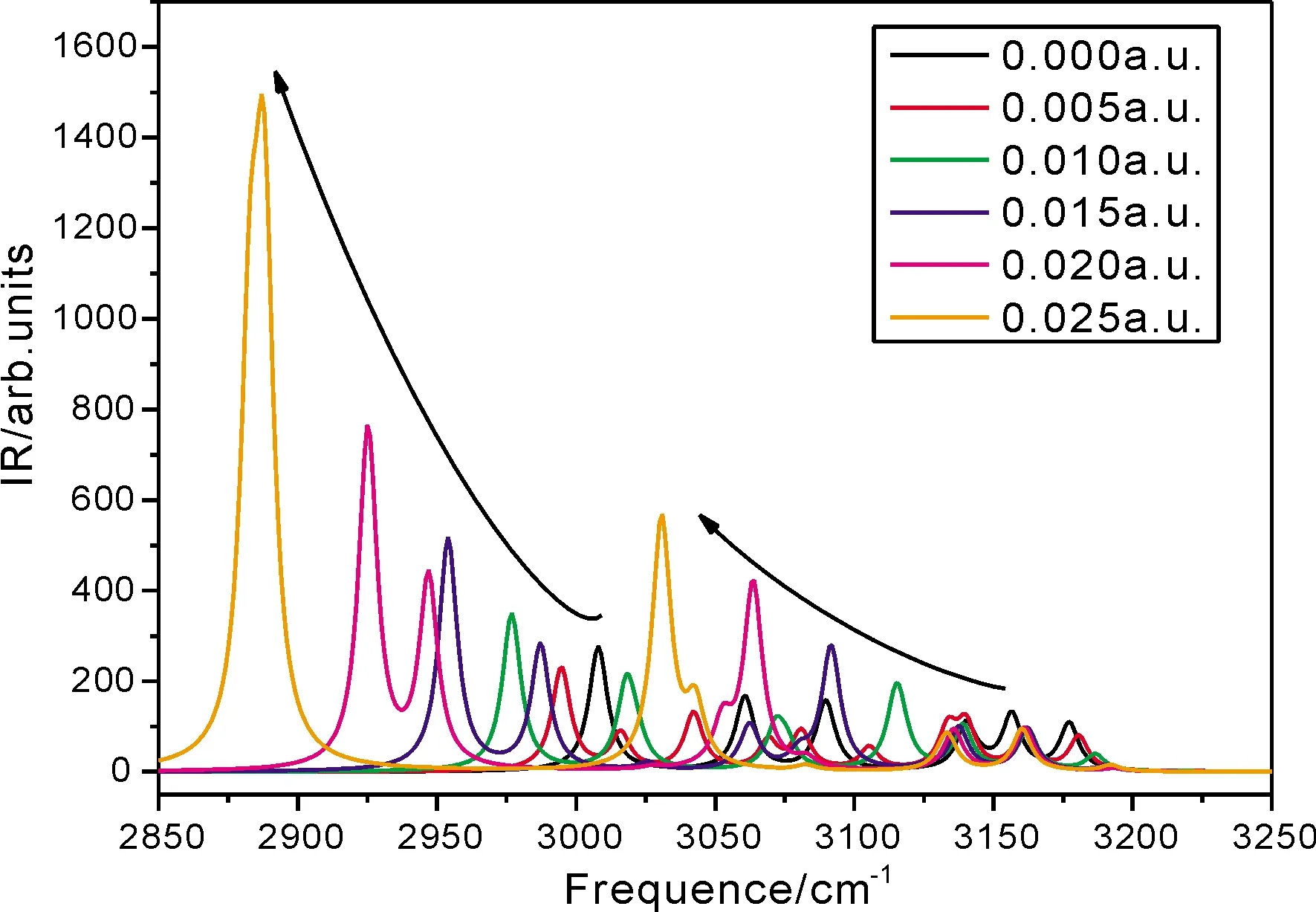
图9 间二甲苯红外光谱随外电场变化趋势Fig.9 The variations of IR spectra of m-xylene under different external field
3.5 外电场对分子解离影响
在两甲基中C原子连线方向施加不同强度的电场( 0 ~ 0.025 a.u.),采用B3LYP/6-311G++方法对间二甲苯分子C6-C11键进行能量扫描,得到了束缚的势能曲线. 如图10所示,随着外加电场强度的增加,C6-C11键势能曲线的束缚状态逐渐被解开,解离的势垒越来越小,C6-C11键越来越容易碎裂. 可以预见,当外加电场强度增加到一定程度时,C6-C11键的势能曲线束缚状态完全消失,解离的势垒完全消失,此时C6-C11键发生降解. 在初步计算过程中,我们发现C6-C11键解离势垒与外加电场强度是呈线性的. 在这里,我们采用线性拟合的方法对C6-C11键解离势垒与电场强度进行拟合,拟合结果展示于图11. 如图11所示,当外加电场约为0.047 a.u.时,C6-C11键解离势垒完全消失,C6-C11键发生断裂. 同时该线性拟合结果的线性相关系数R2为0.9975,十分接近于1,说明势垒与电场强度具有极高的相关性,选用线性拟合得到的结果具有极高精确性. 可以为采用外电场使间二甲苯分子发生降解提供重要参考依据.

图10 间二甲苯分子C6-C11键势能面随外电场变化Fig. 10 The variations of dissociation potential energy surfaces along C6-C11 bond under different external fields
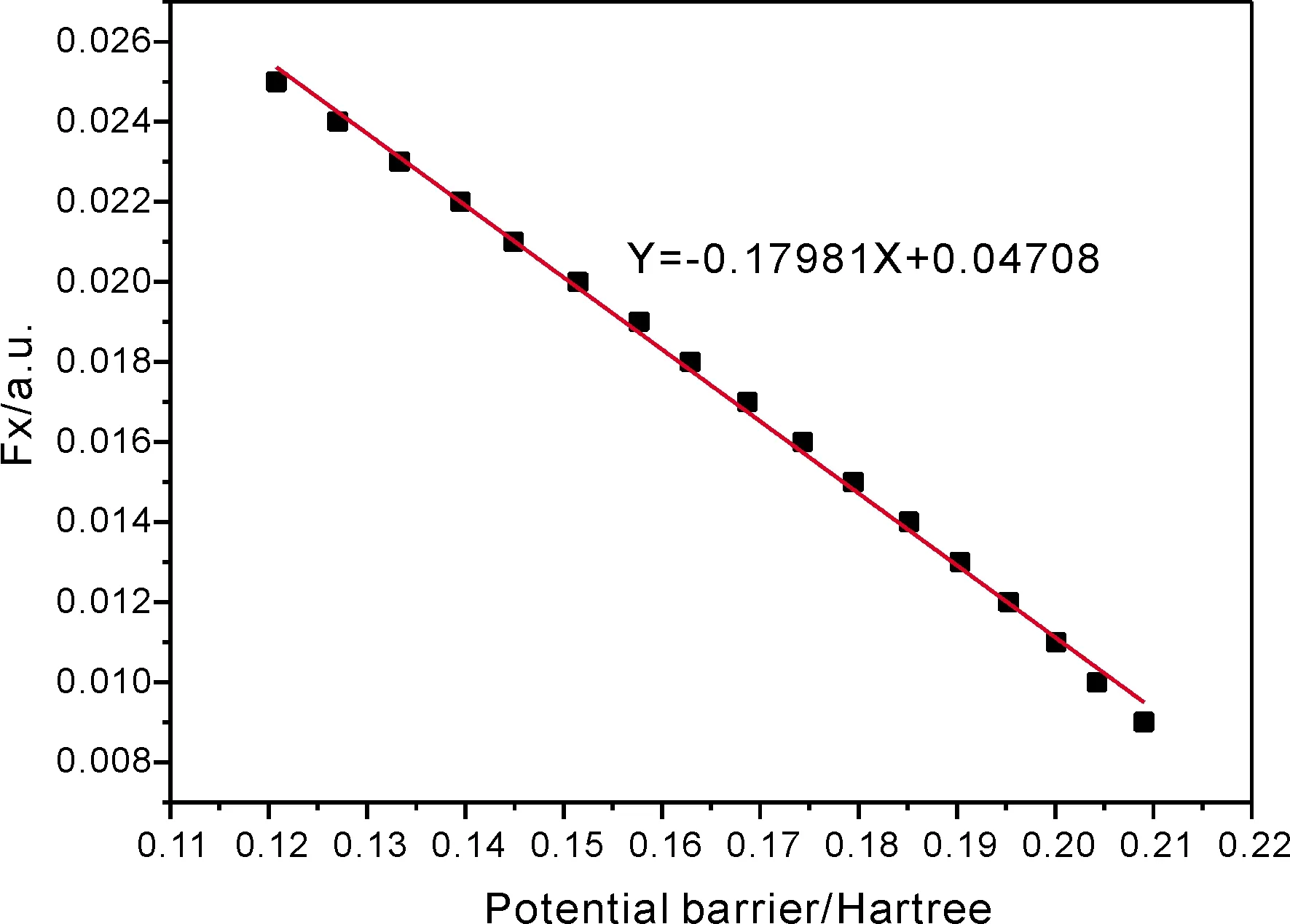
图11 电场强度与C6-C11键解离势垒线性拟合结果Fig. 11 The linear fitting result of electric field intensity with potential barrier of C6-C11`s dissociation
4 结 论
本文计算了间二甲苯分子在外电场下的结构,红外光谱和解离特性. 通过不同的基组计算得到了间二甲苯分子在该基组下的稳定构型,通过对比红外光谱发现,B3LYP/6-311G++基组得到的计算结果与实验值最为接近. 在该基组水平上,我们在两甲基中C原子连线方向施加不同强度的电场,计算出了间二甲苯分子在不同外电场下的分子稳定构型,红外光谱与解离势能面. 研究表明:当沿两甲基C原子连线方向施加的不同强度的电场(-0.025 a.u. ~ 0.025 a.u.)时,分子总能量先增大后减小,C2-C15键变短,越来越稳定. C6-C11键伸长,越容易断裂. C1-C6键先缩短然后几乎没有变化. 电偶极矩先减小后变大,且在无外电场时电偶极矩最小. 能隙先增大后减小,无外电场时能隙最大,说明外加电场后分子更易被激发到激发态来参与或发生化学反应. 同时随着电场强度增加,最低空轨道能量LUMO先增大后减小,分子得到电子的能力先减弱后增强. 随着俩甲基团C原子连线方向的电场强度的增加(0 ~ 0.025 a.u.),C11甲基团对称伸缩振动和C3-C8与C5-C10键伸缩振动均呈现红移,C11甲基团振动强度先减小后变大,而C3-C8与C5-C10键伸缩振动强度单调增加. 随着沿两甲基团中C原子连线方向的正向电场强度的增加,C6-C11键束缚的势能曲线逐渐解开,解离的势垒减小,当电场强度约为0.047 a.u. 附近时,C6-C11键可被降解.
猜你喜欢
分子催化(2022年1期)2022-11-02 07:10:56
化学工业与工程(2022年1期)2022-03-29 01:14:38
中学生数理化(高中版.高考理化)(2021年11期)2022-01-18 05:46:08
火工品(2019年6期)2019-06-05 02:35:44
厦门大学学报(自然科学版)(2018年2期)2018-04-11 07:07:35
新高考·高一物理(2016年7期)2017-01-23 02:52:50
中国塑料(2016年2期)2016-06-15 20:30:00
中学生数理化·高二版(2016年9期)2016-05-14 13:19:35
当代化工研究(2016年5期)2016-03-20 16:21:32
应用化工(2014年11期)2014-08-16 15:59:13
