
固相萃取小柱-超高效液相串联质谱测定牛奶中氯霉素
2020-04-25 12:18:42马晓年梁志坚李怡
中国乳品工业 2020年3期
马晓年,梁志坚,李怡
(昆明市疾病预防控制中心,昆明650228)
0 引 言
牛奶中含有丰富的蛋白质、脂肪、维生素和矿物质等营养物质。2008 年1 月15 日,卫生部发布的《中国居民膳食指南(2007)》介绍了新版《膳食宝塔》凸显牛奶价值,牛奶是摄取营养元素的催化剂以及健康的关键,是健康膳食的基石[1]。但由于奶牛饲养过程中,对泌乳期奶牛用药不当、违反国家规定把抗生素作为奶牛饲料添加剂使用、未经彻底清洗和消毒患病奶牛使用过的挤奶工具及贮奶设备、高温季节掺杂抗生素防止牛奶酸败等原因,造成牛奶抗生素污染[2-3]。牛奶中的抗生素残留主要以氯霉素为主,氯霉素(Chloram⁃phnicol,CAP)属广谱抗生素,能抑制细菌蛋白质的形成,广泛用于动物各种传染性疾病的治疗,特别是用于奶牛乳腺炎的治疗,从而易使奶及奶制品中存在氯霉素残留。由于氯霉素可引起再生障碍性贫血和粒状白细胞缺乏症等疾病[4],1999 年9 月13 日中华人民共和国农业部发布了《动物性食品中兽药最高残留限量》[5]的通知,规定了氯霉素在所有食品动物的可食用组织不得检出。
目前,氯霉素主要通过固相萃取小柱进行前处理的提取、净化[6-7],测定方法有液相色谱[8]、液相色谱-串联质谱[9-15]。液相色谱采用保留时间定性,易出现假阳性,而液相色谱-串联质谱采用核质比定性,具有更高的准确度。液相色谱-串联质谱较于液相色谱具有更高的灵敏度,实现对痕量物质更低的检出限。前处理部分,国标方法[11]采用C18 固相萃取小柱对牛奶进行提取、净化,风险监测工作手册[12]采用MSC 固相萃取小柱提取、净化鸡蛋等高蛋白样品。依据上述信息,本实验对比了C18、MSC 两种固相萃取小柱对牛奶样品中氯霉素的提取效果,为牛奶中氯霉素的测定提供参考依据。
1 材料与方法
1.1 仪器与试剂
QTRAP 4500 质谱分析仪(美国AB SCIEX);Ag⁃ilent 1290 高效液相色谱仪(美国安捷伦);高速冷冻离心机(湖南赫西仪器);振荡器(常州金坛恒丰仪器制造有限公司);旋转蒸发仪(德国IKA);氮吹仪(Reeko Auto EVA-30);电子天平(Sartorius,0.01 mg);均质器;Agela Technologies Cleanert MCS-SPE固相萃取小柱(500 mg/6 mL);Agela Technologies Cleanert S C18-SPE固相萃取小柱(500 mg/6 mL)。
液态牛奶,市购;氯霉素(坛墨质检-国家标准物质中心);氯霉素-D5(坛墨质检-国家标准物质中心);甲醇、乙酸乙酯(色谱纯,美国JT Baker);氨水、氯化钠(分析纯);正己烷(优级纯);实验用水为超纯水。
1.2 方法
1.2.1 标准溶液配制
准确量取氯霉素标准溶液25 μL,用甲醇稀释并定容至100 mL,得到25.00 ng/mL 的氯霉素应用液。准确量取氯霉素-D5 标准溶液20 μL,用甲醇稀释并定容至100 mL,得到20.00 ng/mL 的氯霉素-D5 应用液。标准应用液均于4 ℃条件下保存。
1.2.2 样品的提取
取10 g(精确至0.01g)均质好的牛奶样品于50 mL离心管中,加氯霉素-D5 标准应用液100 μL,再加乙酸乙酯20 mL,振荡15 min,5 000 r/min 离心10 min,取乙酸乙酯层于鸡心瓶中。再加20 mL 乙酸乙酯重复提取一次,合并两次提取液,于40 ℃水浴旋转蒸发至干。用5 mL 4%氯化钠溶液溶解残留物,加5 mL 正己烷振荡混合1 min,静置分层,弃去正己烷。再加正己烷5 mL,重复提取一次,取下层液备用。
1.2.3 样品的净化
C18及MCS小柱依次用5 mL甲醇、5 mL水活化,取备用液分别过C18 及MCS 固相萃取小柱,用5 mL水淋洗抽干,再用5 mL 甲醇洗脱,收集洗脱液于40 ℃下氮气吹干。用1.0 mL甲醇溶解残留物,涡旋混匀,过0.22 μm 滤膜,供超高效液相-串联质谱(UPLC-MS/MS)测定。
1.2.4 仪器条件
色谱:Syncronis C18 色谱柱(100×2.1 mm,1.7 μm);流动相A:0.05%氨水,流动相B:乙腈,梯度洗脱程序件(见表1);流速:0.40 mL/min,分析时间5 min,进样体积5 μL,柱温30 ℃。

表1 流动相梯度洗脱
质谱:离子源:电喷雾离子源(ESI);扫描方式:负离子模式;检测方式:多反应监测(MRM);电离电压:4.0 kV;检测方式:多反应检测(MRM);离子源温度:500 ℃;喷雾器压力:50 psi;辅助加热气:50 psi;气帘气压力:35 psi;其他参数见表2。
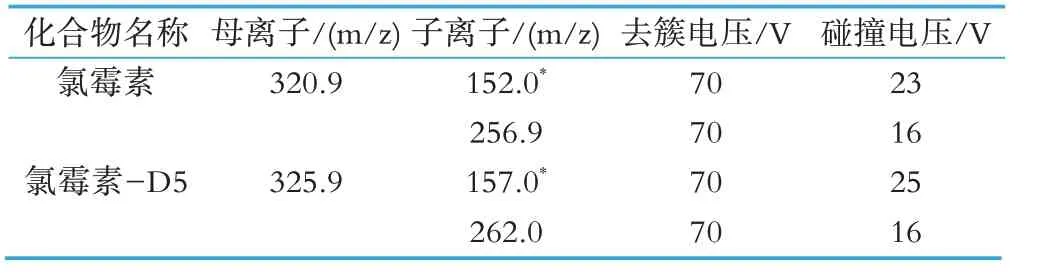
表2 各化合物质谱参数
2 结果与分析
2.1 质谱条件优化
根据待测物的性质,使用氯霉素及氯霉素-D5 标准应用液在ESI-模式下分别进行质谱条件优化。选择每种药物响应强度最高的m/z 值作为母离子,进行子离子扫描,并优化去簇电压和碰撞电压。并在MRM 模式下优化了气帘气、离子源温度、喷雾气、辅助加热气。
2.2 色谱条件优化
本文实验了Hypersll GOLD色谱柱(100×2.1 mm,1.9 μm),Syncronis C18 色谱柱(100×2.1 mm,1.7 μm),发现采用Hypersll GOLD 色谱柱(100×2.1 mm,1.9 μm)分离时,色谱峰展宽,响应值偏低。采用Syncronis C18色谱柱(100×2.1 mm,1.7 μm)分离时,色谱峰得到较大改善,峰形收窄,响应增高,基线噪音降低,实现更低检出限。
本文选择流动相时考虑到是ESI-模式,所以使用了0.05%氨水及具有改善峰形作用的乙腈。低流速时,峰形展宽、拖尾,综合分离效果及柱压等因素,最终选择0.4 mL/min 流速。图1 为1.00 ng/mL 的氯霉素标准溶液MRM 谱图。
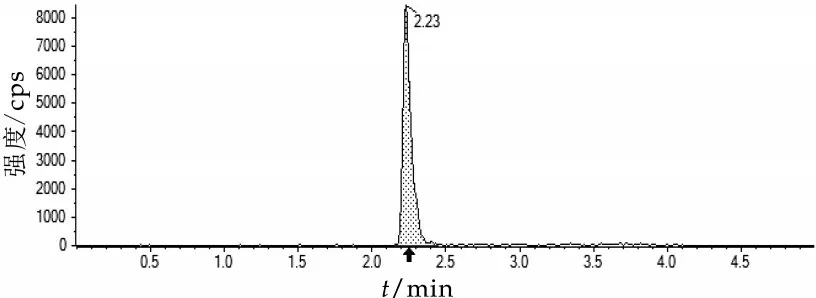
图1 CPA标准MRM谱图
2.3 样品前处理方法的优化
2.3.1 提取溶剂的选择
本文试验了乙腈-乙酸乙酯(10:90)、乙腈-乙酸乙酯(20:80)、乙腈-乙酸乙酯(50:50)及乙酸乙酯的提取效果。乙腈-乙酸乙酯(10:90)、乙腈-乙酸乙酯(20:80)分层效果不理想,震摇乳化。乙腈-乙酸乙酯(50:50)提取,分层效果较好(分为乙腈层、磷脂层、乙酸乙酯层),但加标回收率较低,在21.6%~48.5%之间。乙酸乙酯,振荡提取效果较好,回收率均能达50%以上。
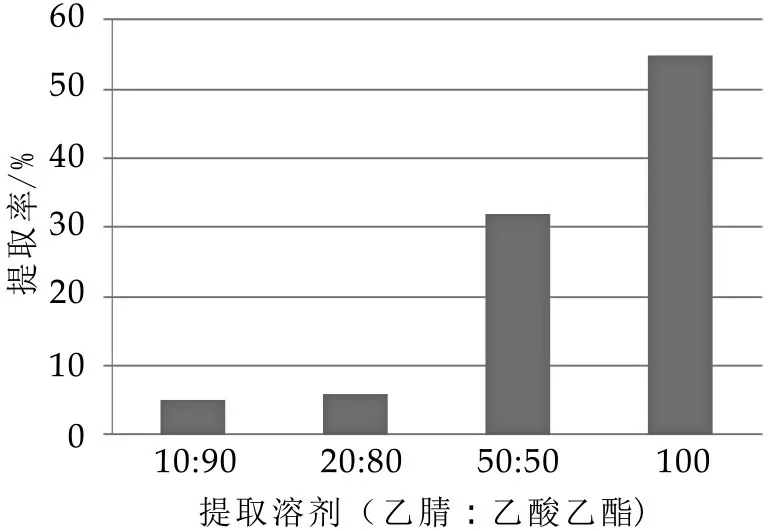
图2 不同提取溶剂对比
2.3.2 提取方法的选择
本文比较了《2019 年国家食品污染物和有害因素风险监测工作手册》[11]中用乙酸乙酯提取一次,氮气吹干水溶解过固相萃取小柱提取、净化,及GB 29688-2013《牛奶中氯霉素残留量的测定液相色谱-串联质谱法》[12]中乙酸乙酯提取两次,旋转蒸发至干,水溶液溶解过固相萃取小柱提取、净化。实验发现,手册的提取方法损失较大,回收率均于60%以下,国标方法提取效果较佳。
2.3.3 固相萃取柱的选择
本文进行了0.10、0.50、2.50、10.00 ng 4 个水平的加标回收实验,比较了C18 及MCS 两种固相萃取小柱对氯霉素的保留能力。结果表明MCS 固相萃取小柱4 个水平的回收均实现75%以上,而C18 固相萃取小柱在低水平回收实验中效果不佳,回收率低于50%。实验结果见表3。
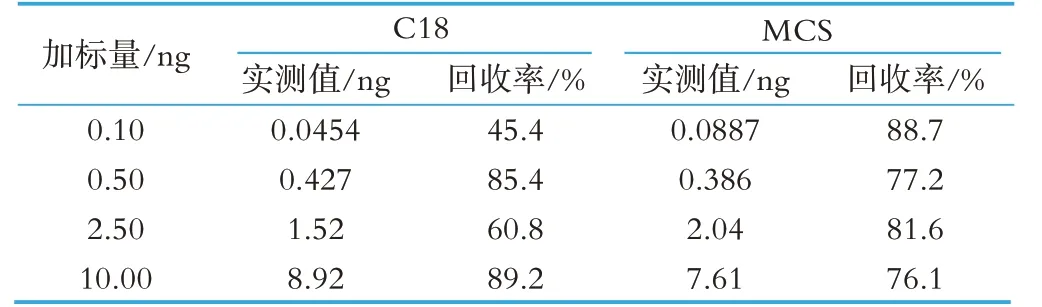
表3 两种固相萃取柱的回收率(%)
2.4 标准曲线及检出限
用甲醇将氯霉素-D5 及氯霉素标准应用液制备成含氯霉素-D5 2.00 ng/mL,含氯霉素分别为0.01、0.05、0.10、0.50、1.00、2.50、5.00、10.00 ng/mL 的标准工作溶液,在确定的分析条件下进行测定,以标准溶液质量浓度为横坐标,氯霉素与氯霉素-D5 响应值之比为纵坐标绘制标准曲线,得到的线性方程、相关系数及检出限见表4。

表4 氯霉素的线性方程、相关系数、检出限
2.5 精密度及准确度

图3 工作曲线
取不含氯霉素的牛奶样品分别过C18 及MCS 固相萃取小柱进行三水平加标回收率实验,结果见表5。由表中可见,C18 固相萃取小柱回收率在31.4%~155%之间,在低水平及高水平的回收率均不理想,而MCS 固相萃取小柱在三个水平的加标回收率均能达到50.4%以上。结合上述固相萃取小柱回收试验,说明MCS 固相萃取小柱有更良好的准确性,适宜于牛奶中氯霉素残留的痕量测定。图4 为牛奶样品添加氯霉素的MRM 谱图。

表5 氯霉素在不同加标水平下的加标回收率及相对标准偏差(n=3)

图4 加标样品MRM谱图
3 结 论
本研究采用2 种不同的固相萃取小柱对牛奶中的氯霉素进行提取、富集和净化,通过对回收率、精密度、定量限等指标进行考察,得出MCS 固相萃取小柱损失小,回收率高,同时检出限可达到0.004 μg/kg,远低于国标方法,且稳定性高,适用于大量样品的集中测定。
建立了UPLC-MS/MS 内标法测定牛奶中氯霉素含量的方法。分析过程中引入氯霉素-D5 作为内标物,对于分析全过程中导致目标物损失给予完全的校正,改善了方法的精密度。以Syncronis C18 色谱柱分离,流动相为0.05%氨水和乙腈,电喷雾负离子MRM 模式检测。该法测定周期短、定性、定量准确,适用于痕量氯霉素的检测。
猜你喜欢
少年文艺(2022年8期)2022-07-08 10:02:47
食品安全导刊(2021年20期)2021-08-30 06:40:34
昆明医科大学学报(2021年8期)2021-08-13 08:59:14
中成药(2018年6期)2018-07-11 03:01:04
中成药(2017年8期)2017-11-22 03:19:25
中国经济周刊(2017年6期)2017-03-21 00:59:27
天然产物研究与开发(2016年11期)2016-06-15 20:29:17
读写算·高年级(2016年3期)2016-05-30 01:53:46
家庭科学·新健康(2016年5期)2016-05-12 23:51:56
中国药物应用与监测(2015年5期)2015-12-11 03:15:53
