
基于异构网络特征与梯度提升决策树的协同药物预测
2020-04-19 07:22聂丽霞
计算机应用与软件 2020年4期
聂丽霞 刘 辉 邹 凌
1(常州大学信息科学与工程学院 江苏 常州 213164)2(常州大学商学院 江苏 常州 213164)3(常州市生物医学信息技术重点实验室 江苏 常州 213164)
0 引 言
因为生物信号通路与蛋白质网络通常存在交互(crosstalk)与补偿性结构,传统的“单药物,单靶标”治疗在复杂疾病特别是癌症的治疗中往往不能有效抑制癌细胞增殖通路,达不到理想的治疗效果[1]。药物组合通过协同干扰生物网络,能更有效地抑制致病基因的活性水平[2]。以往的研究表明[3],与单一药物相比,组合药物能更有效地抑制癌细胞生长或促进癌细胞凋亡,并降低毒性和副作用。但是目前的组合药物发现依赖于临床经验与偶然机会,因此,迫切需要一种理性的、系统的计算方法来筛选组合药物,以减少需要实验验证的候选组合药物数量[4]。
本文构建了药物-蛋白质异构网络,采用重启型随机游走获得组合药物特征,训练梯度提升树算法(Gradient Boosting Decision Tree,GBDT)预测组合药物。本文的贡献在于:1) 使用随机游走从异构网络获得的特征向量相对于本体特征维度大大降低,提高了分类器训练效率,大幅提高了分类器的训练与预测的效率;2) 克服了直接拼接本体属性作为输入特征向量导致特征维度不一致的弊端;3) 显著提高了多种分类器的性能,例如,梯度提升决策树分类器的AUC值从0.528增加到0.909。
1 数据和方法
1.1 数据来源
首先从公共数据资源中收集了包括药物的化学指纹、蛋白质序列、药物-蛋白质关联以及已知药物组合。药物组合来源是DCDB数据库[5],其包含1 363种药物组合(330种批准和1 033种研究,包括237种不成功的用法),涉及904种个体药物和805种目标。训练集的正样本由DCDB中批准和研究的组合药物组成,负样本通过计算机随机产生。考虑到目前非有效药物组合的数量实际上是巨大的,并且远远超过有效药物组合,通过从药物集合组中随机挑选成对药物来产生许多药物组合作为负样本是合理的,实际上该方法广泛应用于预测药物-靶标相互作用和药物-疾病关联研究[6]。
1.2 方法概述
使用药物和蛋白质本体特征计算药物-药物相似度,蛋白质-蛋白质相似度,结合已知药物-蛋白质关联网络,构建了药物-蛋白质异构网络。针对每种组合药物样本,在所构建的异构网络上进行重启型随机游走[7]。当随机游走达到稳定状态时,所得到的概率分布作为该药物组合的特征向量。基于药物组合的特征向量,构建梯度树决策提升(GTDB)分类器以预测新的药物组合。
1.2.1蛋白质-药物关联网络
从STITCH数据库中抽取药物-蛋白质关联,STITCH这是一个综合药物-靶标作用数据库,它收集了来自于生化实验、外部数据库、文献挖掘和计算预测等化合物-蛋白质等相互作用。对于每种相互作用STITCH计算了范围从0到1 000的得分。首先使用阈值0.5(对应于STITCH的500)来过滤掉可信度较低的药物-蛋白质互作用,再针对每种药物选取top 3靶蛋白。如果一种药物结合度得分高于0.5的靶蛋白质不超过3种,那么只考虑这些目标,一共得到8 893个药物-蛋白质关联。假设D=(d1,d2,…,dn)和P=(p1,p2,…,pm)表示药物和蛋白质节点集,A表示药物-蛋白质关联矩阵,如果药物i和蛋白质j之间存在有效的相互作用,则aij为STITCH结合度得分,否则aij=0。
1.2.2药物-药物相似网络
为了扩展DCDB数据库的药物,根据STITCH药物-药物相似度分数,对每种药物选取了top 10最相似的药物来扩展药物的种类[8]。去除重复药物后,获得了3 378种不同的药物。选择类似药物的出发点在于类似药物可能与类似的靶蛋白相互作用,在细胞微环境中发挥相似的治疗功效。这样能够通过向已知的药物及其靶标网络进行推理学习,发现新的药物组合。
本文使用药物的化学指纹来计算每对药物的相似度。使用PaDEL软件[9]生成每种药物的PubChem化学指纹(共880种指纹)。因此,每种药物由880维的二元向量表示,如果药物含有相应的化学指纹,则对应元素为1,否则为0。基于指纹向量计算Jaccard评分作为每对药物化学相似度的指标。Jaccard得分是一种广泛使用的相似性度量,通常被定义为两个样本交集的势除以两个样本集的并集的势。设di和dj为药物di和药物dj的化学指纹向量。Jaccard评分定义如下:
(1)
1.2.3蛋白质-蛋白质相似网络
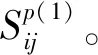
1.2.4异构网络上的重启型随机游走
将药物-药物相似性网络、蛋白质-蛋白质相似性网络和药物-蛋白质关联网络相结合,建立了药物-蛋白质异构网络G=(V,E)。节点集V={D,P}是药物和蛋白质节点集合,边集E={Edc∪Ecd∪Edd∪Ecc}其中Ecc、Edd、Ecd分别是药物-药物关联,蛋白质-蛋白质关联、药物-蛋白质关联的集合,Edc是Ecd的转置矩阵。对于每种组合药物,在异构网络上进行重启型随机游走,模拟组合药物对靶标干扰在蛋白质网络中的扩散效应。当随机游走达到稳态时,游走子的概率分布相当于组合药物对每种蛋白质的干扰强度。数学形式上,对于由两种药物组成的药物组合,使用这两种药物及其已知的靶蛋白质作为种子节点在异构网络上进行重启型随机游走,如图1所示。当随机游走过程达到稳定状态时,此时的概率分布代表组合药物对蛋白质网络的扰动。
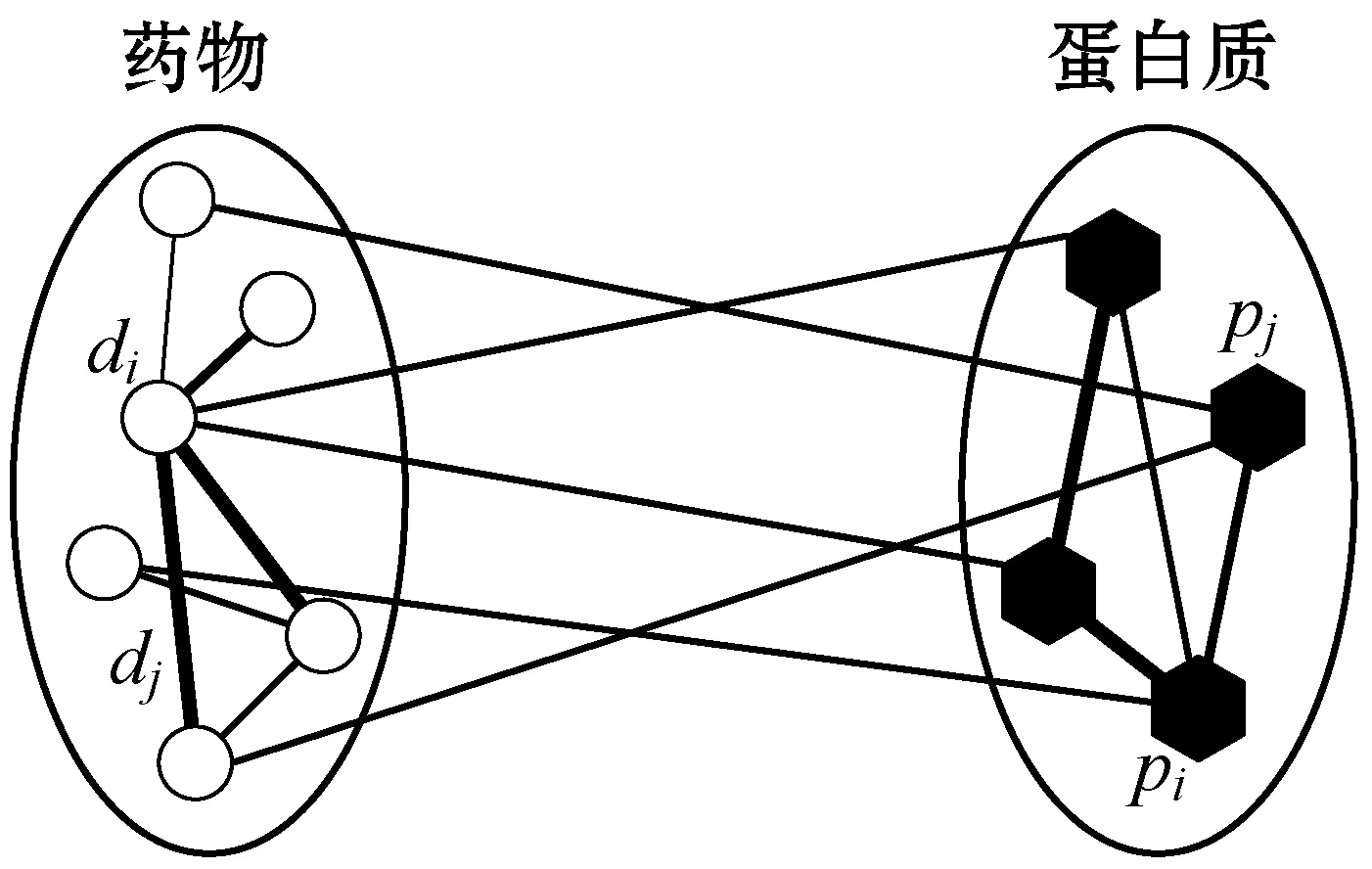
图1 药物-蛋白质异构网络上的重启型随机游走示意图
利用药物-蛋白质异构网络,构建了异构网络的转移矩阵T:
(2)
式中:Tdd和Tpp分别是随机游走过程中从药物节点(蛋白质)到药物节点(蛋白质节点)的概率转换矩阵;Tdp是从药物节点到蛋白质节点的概率转换矩阵;Tpd是从蛋白质节点到药物节点的概率转换矩阵。
假设随机游走子从异质网络中的药物节点开始,以概率λ访问其中之一的目标蛋白质,或以概率(1-λ)访问任何其他药物节点。 如果λ=0,随机游走者只能保持在一种类型的网络中。基于药物-药物相似性,将药物di和药物dj的转变概率定义如下:
(3)
类似地,从蛋白质pi到蛋白质pj的转换概率可以使用如下的蛋白质-蛋白质相似性来定义:
(4)
从药物di到蛋白质pj的转变概率定义为:
(5)
从蛋白质pi到药物dj的转变概率定义为:
(6)
令P(t)为(n+m)维度向量,其中第i个元素是在第t步游走子访问节点i的概率,随机游走过程可以迭代计算:
P(t+1)=(1-α)T′P(t)+αP0
(7)
式中:α是重启概率;P0是由组合药物及其靶蛋白组成的一组种子节点上的初始概率分布。
实际上,药物组合的单药种类不限于两种,因此初始概率分布可以很容易地扩展到多药组合。需要注意的是,采用药物-蛋白质异构网络上随机游走算法的原因在于,该算法能有效地模拟药物分子作用的影响在蛋白质网络中的传播扩散过程,即药物分子在体内与靶标结合之后,抑制或激活靶蛋白的功能,从而引发一系列的级联生化反应。实际上,该算法已经被多项研究使用并被证明是有效的[6,12]。
1.2.5训练梯度提升决策树模型
梯度提升决策树算法(GBDT)是一种有效的机器学习方法,在分类和回归问题上都取得了理想的性能。实际上,Caruana和Niculescu-Mizil对提升决策树算法和其他七种典型分类器进行了综合性能评估,结果表明,基于梯度提升树算法在预测中获得了最佳性能。另一项实证绩效评估也表明,当维数不超过4 000时,梯度提升决策树的表现非常好。因此,使用药物-蛋白质异构网络上随机游走获得的药物组合特征向量来训练GBDT分类器模型预测新的组合药物。
数学形式上,GBDT的决策功能初始化为:
(8)
式中:N是训练集中包含的药物组合的数量。梯度提升树算法重复构造K棵不同的分类树h(x,a1),h(x,a2),…,h(x,aK),每棵分类树都是基于随机选择的子集进行训练的样本,然后迭代地构造加法函数θk(x):
θk(x)=θk-1(x)+bkh(x;ak)
(9)
式中:bk和ak是第k棵分类树h(x;ak)的权重和参数向量。损失函数定义为:
L(y,θ(x))=log(1+exp(-yθ(x)))
(10)
式中:y是真实的类标签;θ(x)是决策函数。通过网格搜索迭代地优化bk和ak,使得损失函数L(y,θ(x))最小化。
本文算法的框架如图2所示,异构网络上的随机游走算法使用R语言实现,梯度提升决策树算法使用Python语言实现,随机游走产生的特征向量加上类标号之后,作为训练梯度提升决策树的训练集。对于算法中包含的超参数,采用网格搜索方法,在基准数据集上进行10倍交叉验证以寻找最佳值。
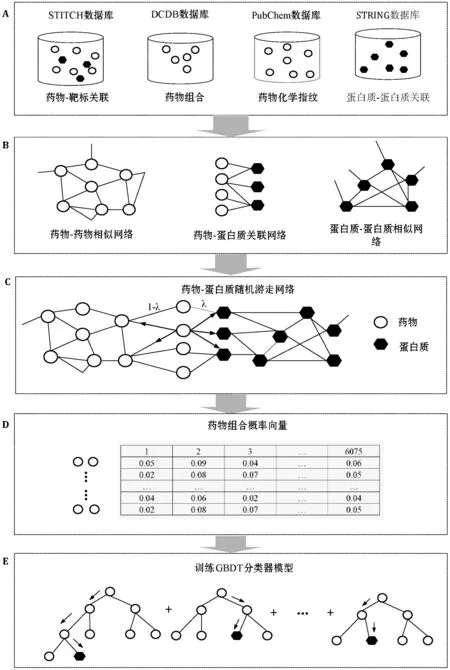
图2 本文算法框架
2 实验评价
2.1 实验方法与性能指标
采用10倍交叉验证对方法性能进行评估,把训练集随机地划分为10个大致相等的子集。轮流使用1个子集作为测试集,其余9个子集作为训练集。该验证过程重复10次,取10次的平均值上作为性能评估指标。实验中所用的性能评估指标包括精度(PRE)、召回率(REC)、F-measure、Matthews相关系数(MCC)和ROC曲线下面积(AUC)。
2.2 与典型分类器的性能比较
为了验证GBDT算法优于其他传统分类器,将其与七种典型分类器包括K最近邻分类算法(KNN)、支持向量机(SVM)、线性回归分析(Logistic)、朴素贝叶斯(Naïve Bayes)、随机森林(Random forest)、Adaboost与LogitBoost的性能进行比较,在DCDB数据集进行了性能评估。对于每种分类器,全部调整它们的参数以获得它们的最佳性能,对于KNN算法,调整K参数,包括1、3、5、7;对于正则化Logistic回归,使用网格搜索调整其权重系数c,从0.1到2.0之间以步长为0.1逐渐增加;对于SVM,其调整系数c在2的区间内从1到10变化且尝试了测试不同的核函数,包括线性、多项式、RBF和S形函数;对于Adaboost,将种子从1逐渐增加到10;对于其他基于树的算法,包括Random Forest和logitBoost,树的最大深度数从5逐渐增加到50,间隔为5;对于朴素贝叶斯分类器,采用其默认设置。表1显示了每种方法获得的最佳性能对应的性能指标。显然,GBDT算法在所有性能指标方面都达到了最佳。

表1 GBDT与其他七种典型分类器在DCDB 数据集上的性能比较
2.3 基于异构网络的特征显著提高分类器性能
为了验证药物-蛋白质异构网络上随机游走所获得特征的有效性,在药物与蛋白质本体特征与基于异构网络的特征之间进行了性能比较。药物与蛋白质本体特征包含药物的化学指纹与蛋白质的GO(Gene Ontology)功能注释词条(terms)。然而,由于不同药物组合的单药和靶蛋白的数量不同,直接连接药物化学指纹和蛋白质GO功能注释作为组合药物的特征,会导致特征向量的维度不一致。所以,先取单药化学指纹的并集与药物靶蛋白GO注释的并集,再将这两种本体特征向量的并集进行联接(向量concatenation操作),即能得到维度一致的组合药物的特征。
将每种组合药物的化学指纹和GO注释的并集作为GBDT分类器的输入特征。性能测量结果如表2所示。从中可以发现基于异构网络特征的GBDT分类器的性能远超在本体特征上获得性能。例如,GBDT分类器的AUC值从0.528增加到0.909。对其他七种典型分类器也在两种特征上进行了性能比较,如表2所示,通过异构网络提取的特征也极大地提高了所有这些分类器的性能。
3 结 语
靶向药物已经在癌症治疗中取得了显著疗效,但是由于癌细胞逐渐产生耐药性,使得靶向药物对肿瘤治疗的临床获益受到很大限制。药物组合协同干扰蛋白质网络能更有效地抑制致癌基因的活性水平,在复杂疾病的治疗中发挥越来越重要的作用。本文将药物相似性网络、蛋白质相似性网络和已知的药物-蛋白质关联整合为药物-蛋白质异质网络。使用药物及其靶蛋白作为种子节点在异质网络上运行重启型随机游走算法,将收敛后的概率分布作为每种药物组合的特征向量,训练梯度提升决策树分类器来预测新的药物组合。使用DCDB基准数据集进行了性能评估实验,结果表明本文算法比七种典型分类器和传统的增强算法具有更高的性能。从网络药理学的角度来看,本文算法有效地利用了生物网络中药物靶标网络的拓扑属性和互作用,是一种系统的组合药物预测方法。
猜你喜欢
中国交通信息化(2022年7期)2022-10-27
现代电子技术(2022年15期)2022-07-28
计算技术与自动化(2022年2期)2022-07-04
九江学院学报(自然科学版)(2022年2期)2022-07-02
小学教学研究(2022年5期)2022-04-28
电子产品世界(2022年4期)2022-04-21
计算机系统应用(2021年2期)2021-02-23
华东师范大学学报(自然科学版)(2019年3期)2019-06-24
福建基础教育研究(2019年3期)2019-05-28
福建基础教育研究(2019年11期)2019-05-28
