
亚洲棉短绒突变体纤维发育及其差异基因表达分析
2020-04-16 10:15:20王晓阳王丽媛潘兆娥何守朴龚文芳杜雄明
作物学报 2020年5期
王晓阳 王丽媛 潘兆娥 何守朴 王 骁 龚文芳,3,* 杜雄明,2,*
亚洲棉短绒突变体纤维发育及其差异基因表达分析
王晓阳1王丽媛1潘兆娥1何守朴1王 骁1龚文芳1,3,*杜雄明1,2,*
1中国农业科学院棉花研究所/ 棉花生物学国家重点实验室, 河南安阳 455000;2棉花生物学国家重点实验室郑州研究基地 / 郑州大学, 河南郑州 450001;3中南林业科技大学 / 经济林培育与保护教育部重点实验室, 湖南长沙 410004
棉花纤维是重要的天然纺织材料, 是最长的单细胞, 是研究纤维发育的良好材料。本研究以亚洲棉短绒突变体(FZ)及其野生型()为材料, 结合扫描电镜、石蜡切片、RNA-seq技术, 解析棉花短绒起始的可能机制。与野生型()的0DPA时期胚珠相比, 突变体的胚珠在该时期仅有少量的纤维起始。在+3DPA时, 突变体没有短绒细胞起始, 仅有长纤维细胞, 而野生型有大量的短纤维细胞和长纤维细胞。对这2个材料的0DPA、+3DPA、+5DPA和+8DPA胚珠差异基因分析结果显示, 在短绒突变体(FZ)和野生型()的4个纤维发育时期共挖掘出3780个差异表达基因, 其中0DPA时差异基因数目最少, 随着胚珠发育时间的延长, 差异基因的数目逐渐增加。KEGG分析发现这些基因主要参与蜡质、角质生物合成, 以及苯丙烷代谢和植物信号传导过程。共表达趋势分析显示, 在突变体+3DPA上调的差异基因中, 参与离子结合、MAPK级联反应、氧化还原活性和转录调控的基因表达受到正影响(表达水平提高), 造成突变体短绒纤维不能正常起始。这些结果描述了二倍体亚洲棉短绒起始的动态变化, 可为进一步研究棉纤维发育提供参考。
亚洲棉; 短绒突变体; 差异表达基因; 共表达趋势
棉花是一种重要的经济作物, 种子是棉籽油的主要原料, 纤维更是一种重要的天然纺织资源。棉花纤维是高等植物中最长的单细胞突起, 由棉花胚珠表皮细胞产生。棉纤维的长度一般为30~40 mm[1]。棉花纤维根据形态可以分为长纤维和短绒2种不同的类型。具有重要经济价值的长纤维在开花前0DPA (day post anthesis)发育, 经过快速伸长, 纤维素生物合成和成熟, 最终形成成熟的长纤维, 通过轧花从种皮上收获并用于纺织产品[2-3]。短绒纤维起始于+3 ~ +5DPA, 长度不到5 mm, 轧花后不能从种皮上分离下来, 紧紧附着于种皮上, 对种子传播起重要作用[4]。胚珠上短纤维细胞和长纤维细胞的共存现象长期以来一直受到研究者的关注, 为研究植物细胞的分化提供了独特的模式体系。
棉纤维的发育受一系列基因调控, 这些基因与植物激素信号转导、细胞能量代谢、脂肪酸代谢、次生代谢等途径有关。目前关于调控纤维发育的基因和调控网络的信息还不太清楚。在棉花基因组测序之前, 许多研究者主要通过基因芯片技术筛选与棉花纤维发育有关的基因, 在分子水平上了解纤维发育的机制。如Shi等[5]通过cDNAs芯片分析技术发现了778个cDNAs优先在发育的纤维中表达。同年, Yang等[6]通过分析陆地棉及其光子突变体转录组, 鉴定出了受激素诱导的转录因子、、在纤维起始过程中高表达。随着棉花基因组测序的完成, 逐步完善了大量的棉花转基因技术, 使得通过转录组分析与纤维发育相关的基因与代谢通路成为可能[7-9]。鉴定出一些与纤维发育及调控细胞壁沉积与强度的候选基因[10]。通过分析0DPA和+10DPA的二倍体亚洲棉近等基因系有短绒无长纤维材料()和既有短绒又有长纤维材料(FL)的转录组发现, 一些参与植物激素信号转导、细胞能量代谢, 脂肪酸代谢、刺激代谢的转录因子如、、、、、、、、、和在长纤维突变体+ 10DPA纤维中明显下调, 植物受体样激酶(plant receptor-like kinases, RLKs)、富含亮氨酸(Leucine Rich Repeats)的LRR家族蛋白和参与信号转导的编码丝裂原活化蛋白激酶(mitogen-activated protein kinase, MAPK)在突变体中也下调, 导致纤维生长减慢[11]。
虽然上述转录组分析的研究为棉纤维的起始过程提供了一些研究基础, 但纤维发育的关键基因的研究仍处于初级阶段, 长纤维和短绒纤维发育及生理过程仍然不清楚, 并且目前这些转录组研究主要集中在纤维伸长阶段, 关于短绒纤维起始的动态过程研究较少。
突变体特有的性状是研究基因表达的有利资源, 分析植物突变体加速了特定基因功能的发现。本研究所用的材料为二倍体亚洲棉短绒突变体GA0149有长纤维没有短绒(其光籽性状受单基因显性控制, 基因型为FZ)及其野生型GA0146既有长纤维又有短绒(基因型为), 经过连续十代的自花授粉, 突变体植株光籽性状稳定, 突变体和野生型材料在形态和发育上区别微乎其微, 除了光籽突变体种皮上没有短绒[8]。利用转录组测序技术分析突变体和野生型0DPA、+3DPA、+5DPA和+8DPA 4个时期的差异基因表达情况, 旨在探究棉花短绒起始的动态过程及其关键基因, 为棉花短绒起始的分子机制提供坚实基础。
1 材料与方法
1.1 试验材料
亚洲棉野生型毛子材料为中美棉DPL971 (), 短绒突变体材料为中美棉DPL972 (FZ), 均来自棉花种质资源中期库近10年的自交材料。2017年在中国农业科学院棉花研究所实验基地, 用标准的田间管理措施, 按3个重复种植每个材料。
1.2 扫描电镜
将2个材料(和FZ) 0DPA、+3DPA的胚珠固定于含有2.5%戊二醛的0.1 mol L–1磷酸缓冲液中, 用真空泵抽真空15 min, 使胚珠完全浸没于固定液中, 4℃过夜后用0.1 mol L–1pH 7.2磷酸缓冲液洗3次, 每次10 min;接着, 用ddH2O清洗2次, 每次30 min; 用30% (v/v)、50% (v/v)、70% (v/v)、80% (v/v)、95% (v/v)乙醇对样品梯度脱水, 每个梯度脱水20 min; 用无水乙醇对样品进行3次脱水; 再用等体积的乙醇: 乙酸异戊酯置换15 min; 最后用100% 的乙酸异戊酯置换15 min; 将样品放于样品笼中用CO2干燥, 然后喷金, 用JSM-6390/LV扫描电镜观察。
1.3 石蜡切片
将新鲜的0DPA、+3DPA的胚珠固定于含有4%的FAA (甲醛-冰乙酸-无水乙醇)固定液中, 抽真空, 保存于4℃冰箱中过夜, 用系列梯度浓度的乙醇(50%、70%、85%、95%和100%)对样品进行脱水处理, 每梯度30 min; 将脱水处理过的胚珠包埋于处理过的石蜡(Sigma-Aldrich)中, 60℃透蜡2 h; 然后将胚珠放于预热的包埋框中, 迅速倒入融化的石蜡, 过夜使石蜡凝固。用石蜡切片机, 切成8 μm 厚的连续切片, 移至42℃的水浴中, 使样品展平, 将切片放在载玻片上, 37℃干燥过夜。用番红固绿染色, 用带有数码相机的显微镜照相。
1.4 文库构建与转录组测序
用北京天根公司的多糖多酚植物总RNA试剂盒提取FZ和0DPA、+3DPA和+5DPA 的胚珠及+8DPA的纤维的RNA。将检测质量合格的RNA送至北京百迈客生物公司进行cDNA文库的构建, 借助 Illumina HiSeq 2000测序仪进行双末端测序, 获得原始数据。
1.5 转录组原始数据的评估以及分析
将得到的原始数据转化为FASTQ形式进行保存, 通过fastp软件过滤掉低质量数据和接头序列, 得到高质量的reads, 即clean data.利用TopHat2/Bowtie2软件将 clean reads 比对到亚洲棉三代基因组上[8,12-13]。同时对比对到参考基因组的序列进行注释, 在比对过程中, 每个错配最多允许2个碱基。采用FPKM值(Fragments Per Kilobase of transcript per Million fragments mapped)来衡量基因表达[13]。根据FPKM值, 以错误发现率FDR<0.01, |log2fold change| ≥ 1,-value<0.01作为筛选突变体和野生型差异表达基因的标准。将获得的差异基因进行GO功能富集分析(WEGO: http://wego.genomics.org.cn/cgi-bin/wego/ index.pl)和KEGG生物通路分析[14-15]。
1.6 荧光定量Q-PCR验证
通过分析RNA-seq数据, 验证转录组数据的准确性, 随机挑选22个差异表达的基因, 利用qRT-PCR进行验证。利用在线软件NCBI-Primer (http://www.ncbi. nlm.nih.gov/tools/primer-blast/)设计引物(表1)。分别取不同组织样品的总RNA 1mg, 利用cDNA反转录试剂盒PrimeScript RT reagent Kit (Perfect real time TaKaRa生物)合成cDNA。利用北京全世金生物公司荧光定量试剂盒(TransStart Top Green qPCR SuperMix), 以UBQ作为内参基因, 利用ABI7500fast Real-time PCR System (美国, ABI公司)开展荧光定量PCR验证。每个基因设计3次技术重复和3次生物学重复。以材料0DPA样品的基因表达量作为1, 分析突变体和野生型各个时期的基因相对表达量, 按2–ΔΔCT法计算相对表达量[16]。
2 结果与分析
2.1 二倍体亚洲棉短绒纤维起始的动态学观察
前人研究结果表明, 长纤维细胞起始于开花当天(0DPA), 在纤维发育早期阶段如在+3DPA时, 长纤维已经突破表皮细胞, 长度约为2 mm, 而短纤维细胞还未突破表皮, 但是胚珠表皮细胞开始出现排列不规则现象。为了鉴定野生型毛子和突变体FZ短绒纤维起始的差异, 取这2个材料的0和+3DPA的胚珠, 用扫描电镜观察发现, 在0DPA时野生型和突变体都有明显的纤维突起, 但是野生型纤维明显长于突变体, 而且纤维数目多于突变体; 当胚珠发育到+3DPA时,和FZ长纤维长度基本一致, 但纤维数目多于FZ, 由于胚珠表面长纤维的干扰, 无法观察到胚珠表面的短纤维(图1-A)。为了进一步验证短绒在细胞水平上的突起, 在电子显微镜下观察和FZ 0DPA和+3DPA的胚珠的石蜡切片, 如图1-B所示,在野生型胚珠表面能观察到长纤维和短纤维细胞, 其中红色箭头所指位置为长纤维, 而黑色箭头所指位置为短纤维细胞。在0DPA时野生型纤维细胞开始隆起, 而突变体只有少量突起; 随着胚珠的发育和 FZ 突起细胞数目都有所增加, 但是在+3DPA时, 短绒纤维开始在胚珠表面出现, 而 FZ 胚珠在相同的发育阶段没有短绒细胞突起, 表明短纤维细胞在FZ中起始失败。
2.2 转录组数据质量评估
为了进一步理解短绒发育的动态过程和鉴定与短绒发育相关的基因, 选取突变体FZ和野生型材料4个发育时期的胚珠和纤维(0DPA、+3DPA、+5 DPA、+8DPA)的RNA, 每个材料3个生物学重复, 构建了24个RNA文库。
24个测序文库获得的约3.2亿条原始数据经去接头和过滤后, 产生出约1.6亿条clean reads。每个文库平均得到65,841,318个高质量的reads (表2)。大约有88%的clean reads可成功比对到已发表的亚洲棉三代基因组上。比对到参考基因组上唯一位置和多个位置的比例分别约为76%~85%、3.53%~6.00%。比对到基因组正链的数目与比对到负链的数目几乎相当。另外每个样本的Q30比例均大于86%, GC含量在35%~65%衡量指标之间。

图1 野生型fz和短绒突变体FZ材料中短纤维发育过程的形态学观察
Fig. 1 Morphology analysis of fuzz-fiber development in normal wilt-type fz (upper) and fuzz mutant FZ (lower)
A: 野生型、短绒突变体的种子以及扫描电镜下看到的短绒纤维起始; B: 石蜡切片所示短绒纤维起始。红色箭头所示长纤维, 黑色箭头所示短纤维细胞。
A: the seeds of wild type, natural fuzz mutant and fuzz fiber initiation under scanning electron microscope; B: paraffin section showing fuzz fiber initiation. Red arrow indicates lint fiber, and black arrow indicates fuzz fiber cells.

图2 所有样本间皮尔逊相关系数分析
A和B分别代表和FZ。A and B indicateand FZ, respectively.
为了鉴定24个样品间的生物学重复关系, 对各样本间生物学重复数据进行皮尔逊相关系数(Pearson Correlation Coefficient, PCC)分析(图2), 结果发现,材料3个生物学重复间的皮尔逊相关系数均大于0.98 (图2-A), FZ 材料3个生物学重复间的皮尔逊相关系数均大于0.94 (图2-B), 说明这2个材料不同样本的3个生物学重复较好, 可进行差异基因表达分析。
2.3 差异基因表达分析
计算FPKM值后, 选择-value<0.05和|log2(ratio)| ≥1作为筛选差异表达基因的标准。通过比较突变体 FZ和野生型材料0、+3、+5、+8DPA的FPKM值, 共鉴定出3784个差异基因(图3-A)。其中在vs. FZ 0DPA时期特异表达的基因有201个,vs. FZ +3DPA时期特异表达的基因有313个,vs. FZ +5DPA特异表达的基因有1108个,vs. FZ +8DPA时期特异表达的基因有1303个。突变体和野生型4个纤维发育时期发现91个共有的差异基因, 这些基因的差异表达情况见图3-B和表3, 52个基因明显上调, 39个基因明显的下调,对差异基因功能分析发现一些未知蛋白、转录因子、乙烯受体和抗病基因都参与了短绒纤维的发育。
通过比较同一材料不同纤维发育时期的差异基因发现(图4-A)在材料中, 开花当天至+3DPA时共有2227个基因上调, 1113个基因下调; +3DPA至+5DPA时期, 601个基因上调, 194个基因下调; +5DPA至+8DPA时期2993个基因上调, 3703个基因下调。+3DPA至+5DPA时期差异基因数目较少。在FZ材料中, 0到+3DPA时期, 2420个基因发生了上调, 下调基因的数目是1201; +3DPA到+5DPA时期, 611个基因上调, 753个基因下调; +5DPA到+8DPA时期, 5465个基因上调, 6686个基因下调。
比较突变体FZ和野生型同一纤维时期的差异基因(图4-A, B)表明, 在开花当天与野生型材料相比,突变体中341个基因上调, 174个基因下调; 在+3DPA时, 突变体中612个基因上调, 下调基因数目170; 在+5DPA时期, 突变体中发生了1018个基因上调, 下调基因数目为802个; 在+8DPA时期, 1230个基因下调, 641个基因上调; 突变体中在短绒起始期上调基因的数目多于下调基因的数目, 这些结果证明大部分基因负调控短绒纤维的起始。
2.4 与短绒起始相关的差异基因KEGG通路分析
通过对2个材料4个纤维发育时期的差异基因KEGG (Kyoto Encyclopedia of Genes and Genomes)通路富集分析(图4-C)发现, 40个基因参与到植物-病原菌互作过程; 20个基因参与到精氨酸和脯氨酸代谢过程; 52个基因参与到内质网上蛋白加工过程; 30个基因涉及到苯丙烷代谢过程; 17个基因涉及到角质、木质素和蜡质生物合成过程; 44个基因参与到苯丙烷生物合成过程; 92个基因汇集到植物激素信号传导过程。对KEGG途径富集因子较高的植物激素信号传导途径中的苯丙烷生物合成途径和角质木质素以及蜡质生物合成途径的差异基因进一步分析发现(表4和表5), 26个差异基因与生长素响应以及生物合成有关, 与野生型()相比, 突变体(FZ)大部分基因在+5DPA时上调, 在+8DPA时下调, 在+3DPA时基因表达差异不明显, 表明生长素(IAA)对短绒的起始没有影响, 对短绒纤维的伸长起正调控作用; 12个基因参与脱落酸(ABA)和乙烯信号转导过程, 如乙烯响应因子和脱落酸受体, 这些基因在短绒纤维起始时期+5DPA突变体中显著上调,基因差异倍数达到15以上, 而这些基因在纤维伸长时期+8DPA突变体中下调, 这些结果说明棉花体内过量的ABA和乙烯抑制纤维的发育, 而少量的乙烯能促进纤维伸长。参与角质、软木质、蜡质生物合成途径的基因有8个, 主要是细胞色素氧化酶P450和脂肪酸羟化酶(表4); 参与苯丙烷代谢和生物合成途径有关的基因有23个, 大部分为过氧化物酶以及2个苯丙氨酸裂合酶、3个甲基转移酶和1个花青素葡萄糖基转移酶, 说明这些基因与短绒纤维的起始和发育有关。

图3 fz及其短绒突变体FZ不同纤维发育时期共有的差异基因
A: 在和FZ材料中不同纤维发育时期差异基因韦恩图; B: 在和FZ两个材料中4个纤维发育时期91个共有的差异基因热图。
: Venn diagram of significant DEGs found inand FZ across various fiber development stages; B: Heatmap showing 91 common significant DEGs inand FZ across fiber development stages.

图4 fz及其短绒突变体FZ不同纤维发育时期差异基因概况
A: 在和FZ材料中不同纤维发育时期差异基因数目; 红色代表上调的基因, 绿色代表下调基因。B: 1783个差异基因在和FZ材料4个短绒纤维发育时期的表达趋势。C:和FZ材料4个短绒纤维发育时期差异基因KEGG 富集通路。
A: number of genes differentially expressed during fiber development within and betweenand FZ; Red represent up-regulated, green represent down-regulated gene. B: the expression trends of 1783 DEGs inand FZ across fiber development stages; C: the DEGs KEGG pathway inand FZ across fiber development stages.

表3 在fz和FZ两个材料中4个纤维发育时期91个共有的差异基因及其表达情况

(续表3)

(续表3)
2.5 差异基因共表达趋势分析
为了研究野生型()和短绒突变体(FZ)的差异基因在胚珠和纤维中的表达情况, 对2个材料4个纤维发育时期的3784个差异基因绘制时空表达模式图(图5-A), 结果发现3784个基因在2个材料4个纤维发育时期可以划分为9组(cluster), 同一组的基因有相同的表达趋势。clusters 1和2的基因在突变体(FZ)的开花当天表达高, 主要参与应答胁迫反应以及能量传递过程; clusters 4、5和7的基因在突变体的+3DPA表达高, 主要参与转录调控和钙离子, 铁离子, 铜离子的结合; clusters 6 和8的基因在突变体材料开花后5 d表达高, 也主要涉及转录调控, 钙离子, 铁离子和铜离子的结合过程; clusters 3和9的基因主要在突变体+8DPA表达高, 主要参与水解酶活性, 转运活性, 核酸激活, 脂肪酸生物合成途径和ATP结合过程(图5-B)。

表4 角质、软木质、蜡质生物合成途径以及苯丙烷代谢途径的差异基因及其表达情况
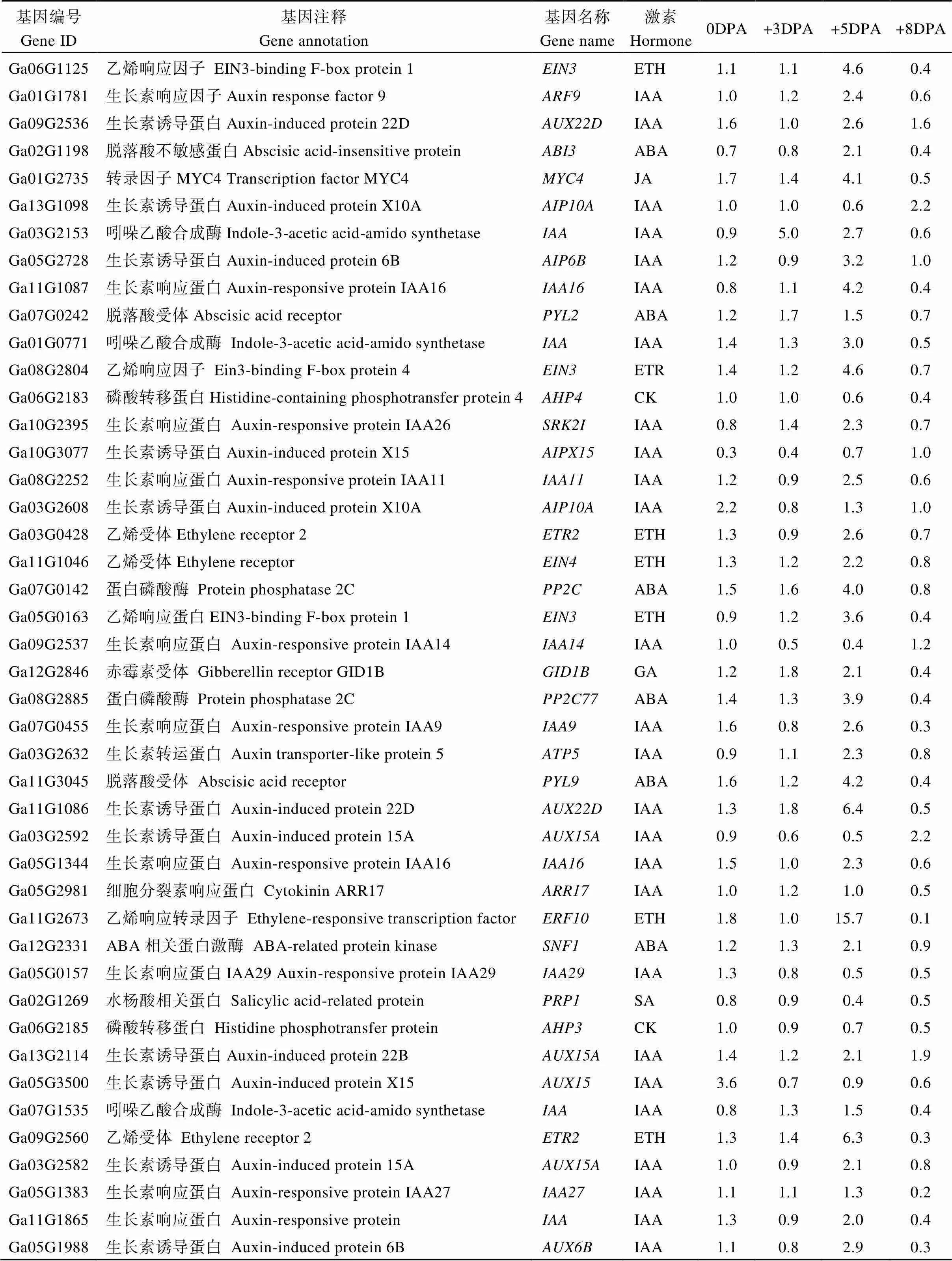
表5 植物激素转导途径中的差异基因及其表达情况

图5 差异基因共表达趋势
A: 在和FZ两个材料中4个纤维发育时期所有差异表达基因的热图分析。B: 差异基因功能注释; 黑色代表0DPA时期高表达的基因, 蓝色代表+3DPA时期高表达的基因, 橙色代表+5DPA时期高表达的基因, 绿色代表+8DPA时期高表达的基因。
A: heatmap analysis of the expression of all significant DEGs betweenand FZ at four time points. B: functional annotations of significant DEGs; Black bars indicate genes highly expressed at 0DPA, blue bars indicate genes highly expressed at +3DPA, orange bars indicate genes highly expressed at +5DPA, and green bars indicate genes highly expressed at +8DPA.
2.6 陆地棉控制短绒基因在亚洲棉短绒突变体FZ及其野生型fz的表达分析
前人通过图位克隆, 在四倍体陆地棉中分别定位出了一个控制短绒发育和长纤维发育的基因, 即MYBMIXTA-like转录因子和;在TM-1短绒起始期表达量明显高于其在短绒突变体中的表达量;在仅有长纤维的短绒突变体纤维起始期的表达量明显高于其在陆无絮中的表达量[17-18]。为了验证和在亚洲棉短绒突变体FZ和其野生型中的表达量, 将这2个基因的蛋白序列在亚洲棉中进行同源性比对(图6)表明,和在亚洲棉中为同一个基因, 基因功能注释显示该基因属于MYB类转录因子, 其相似性达到97%。转录组数据分析表明,在FZ材料短绒起始期表达量高于其在材料中的表达量(图7), 说明可能负调控短绒发育。
2.7 差异基因实时荧光定量PCR验证
为了验证转录组数据的可靠性, 随机选择22个差异基因在2个材料+3DPA胚珠中进行荧光定量PCR验证。结果显示, RNA-seq结果与荧光定量PCR中的基因表达变化一致。通过单样本检验, 野生型+3DPA胚珠中22个差异基因表达量与突变体FZ胚珠中表达量有显著差异。将qRT-PCR和RNA-seq中差异基因表达量经 log2转化后做相关性分析, 所得的皮尔逊相关系数为0.89 (图8), 证明RNA-Seq结果可信。

图6 MYBMIXTA-like基因在亚洲棉中的同源序列比对

图7 Ga12G1199在FZ和fz材料中的表达差异
*< 0.05, **< 0.01, ****< 0.0001.
经qRT-PCR验证, 细胞色素氧化酶P450 ()、甲基转移酶()、转录因子WD40()、蛋白受体激酶()、二氢黄酮醇-4-还原酶()和谷胱甘肽S-转移酶()在突变体FZ中上调; 分子伴侣()、钙联蛋白()、转录因子MYB108()、网状蛋白()和NADH脱氢酶()在突变体材料中下调。
3 讨论
棉纤维起始是决定纤维产量的关键过程。在过去的10年中, 许多参与棉纤维发育的基因已经被鉴定出来。相比之下, 对于野生型种皮上的短绒发育了解比较少。长纤维在生产上主要用于纺织, 短纤维在生产上主要用于火药、造纸和医药用品, 由于短纤维一些特有性质和价值, 吸引了一些科学家研究其发育机制[19]。
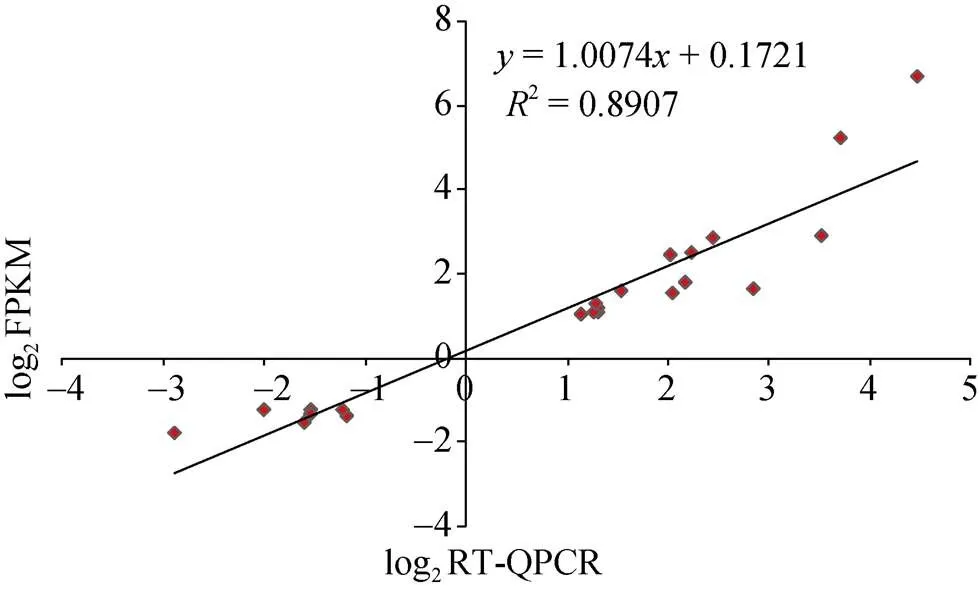
图8 RT-QPCR (X-轴)结果与RNA-seq (Y-轴)差异基因相关性分析
散点图代表RT-QPCR与RNA-seq的差异基因相对表达量的log2值。
Scatter plots represent log2expression ratios calculated from RT- QPCR and RNA-seq.
植物激素例如赤霉素(GA)、生长素、细胞分裂素、油菜素内酯(BR)、脱落酸(ABA)、乙烯、茉莉酸甲酯(JA)和水杨酸是植物内源小信号分子[20]。许多研究证明生长素在纤维发育过程中起重要作用。外源补充吲哚乙酸(IAA)能弥补纤维伸长的缺陷性, 并且能显著增加纤维的总体积[21]。生长素响应蛋白GhARF2和GhARF18, 在纤维起始的细胞中表达高, 当把这2个生长素响应蛋白在拟南芥中超表达后, 能显著促进拟南芥表皮毛起始, 说明GhARF2和GhARF18正调控棉花纤维细胞起始[22]。与和作用相反, INDOLEACETIC()负调控纤维的起始和伸长。在野生型胚珠中表达非常低, 然而在无短绒无长纤维突变体中转录水平在开花后急剧升高[23], 这与本研究结果一致。在短绒突变体+5DPA的表达量比野生型的高4倍。前人研究显示向体外离体培养的胚珠中加入ABA, 纤维细胞的起始受到抑制, 而且突变体0DPA的胚珠中积累ABA的量比野生型的高[24]。这些结果说明ABA负调控棉花纤维起始。本研究结果显示, 与ABA信号传递相关的基因表达量在突变体中明显升高, 与纤维起始呈负相关的植物激素ABA响应因子(、)在突变体短绒起始期也显著的上调, 进而抑制了短绒纤维的起始。这些转录组分析结果需要进一步的试验验证。
最近研究显示, 植物体内信号分子, 如钙离子(Ca2+)和活性氧(ROS), 特别是超氧阴离子(O2-)和过氧化氢(H2O2)参与棉花纤维发育。当用H2O2处理光子突变体()和短绒突变体()时, 突变体纤维在0DPA时开始起始, 说明ROS对纤维起始具有促进作用[25]。本研究显示参与活性氧清除的谷胱甘肽转移酶(GST)基因在突变体FZ短绒起始期明显升高。Ca2+也参与棉纤维细胞的起始, 与–1DPA的胚珠相比, 0DPA的胚珠中钙离子的积累明显升高, 基因芯片分析结果显示, 参与钙离子信号转导的基因钙结合蛋白在+1DPA胚珠中表达量明显上调[26]。钙调蛋白(CBL)在Ca2+信号转导过程中起重要作用, 其表达量在短绒突变体胚珠中下调了2倍, 与前人结果一致。Ca2+正调控纤维起始[27-28]。
前人研究结果显示,()基因正调控陆地棉短绒纤维起始, 而基因正调控陆地棉长纤维起始[17-18]。而这2个基因在亚洲棉中的同源基因在短绒突变体中的表达量明显高于其在野生型中的表达量, 该基因可能负调控短绒的发育。造成这种现象的原因可能是陆地棉和亚洲棉短绒发育机制不一样。
本文通过短绒突变体及其野生型4个纤维发育时期的RNA-Seq测序数据分析筛选到4988个差异表达的基因, 其中上调表达的2612个, 下调表达的2376个, 但仅21.5%的基因在棉花基因组数据库中找到注释信息, 有大量未知基因信息存在。参与KEGG排名前5富集通路的基因, 如参与角质、木质素和蜡质合成过程的基因(, 细胞色素氧化酶)在突变体(FZ)中下调, 说明这些基因可能正调控短绒的发育; 参与苯丙烷代谢和生物合成过程的基因(、, 过氧化物酶)在FZ中明显上调, 说明这些基因可能负调控短绒发育; 参与植物激素信号转导途径中的乙烯响应因子相关基因(、、和)以及ABA合成和信号转导相关基因(、、、和)在FZ短绒起始期+5DPA明显上调, 说明这些基因可能负调控短绒发育。在91个共有的差异基因中,与转座现象相关的基因copia型多蛋白, 转录因子NAC1(), 以及氧化还原相关基因谷胱甘肽S-转移酶()在FZ中明显上调, 说明这些基因可能负调控短绒发育; 而NADPH脱氢酶()基因, 与钙信号转导相关基因()在FZ中下调, 说明这些基因正调控短绒发育。
4 结论
短绒突变体(FZ)及其野生型()的表型差异为阐明调控棉纤维发育的机制和品系间的形态差异提供了信息。短绒的起始在+3DPA。造成短绒纤维细胞在突变体中未起始的原因是, 一些促进纤维伸长的基因下调, 而另外一些抑制纤维起始的基因上调。例如参与蜡质合成的基因以及Ca2+信号转导的基因在突变体中都下调; 清除活性氧的基因以及参与ABA信号转导的基因在突变体中上调。该研究对于理解亚洲棉短绒纤维起始的机制具有一定的理论意义, 对于提高棉纤维产量和品质具有重要的实践意义。
[1] Lee J J, Woodward A W, Chen Z J. Gene expression changes and early events in cotton fibre development.,2007, 100: 1391–1401.
[2] Kim H J, Triplett B A. Cotton fiber growth in planta and. Models for plant cell elongation and cell wall biogenesis., 2001, 127: 1361–1366.
[3] Turley R B, Kloth R H. Identification of a third fuzzless seed locus in upland cotton (L.).,2002, 93: 359–364.
[4] Arpat A B, Waugh M, Sullivan J P, Gonzales M, Frisch D, Main D, Wood T, Leslie A, Wing R A, Wilkins T A. Functional genomics of cell elongation in developing cotton fibers.,2004, 54: 911–929.
[5] Shi Y H, Zhu S W, Mao X Z, Feng J X, Qin Y M, Zhang L, Cheng J, Wei L P. Transcriptome profiling, molecular biological, and physiological studies reveal a major role for ethylene in cotton fiber cell elongation., 2006, 18: 651–664.
[6] Yang S S, Cheung F, Lee J J, Ha M, Wei N E, Sze S H, Stelly D M, Thaxton P, Triplett B, Town C DAccumulation of genome-specific transcripts, transcription factors and phytohormonal regulators during early stages of fiber cell development in allotetraploid cotton.,2006, 47: 761–775.
[7] Wang M J, Tu L L, Yuan D J, Zhu D, Shen C, Li J Y, Liu F Y, Pei L L, Wang P C, Zhao G N,Ye Z X, Huang H, Yan F L, Ma Y Z, Zhang L, Liu M, You J Q, Yang Y C, Liu Z P, Huang F, Li B Q, Qiu P, Zhang Q H, Zhu L F, Jin S X, Yang X Y, Min L, Li G L, Chen L L, Zheng H K, Lindsey K, Lin Z X, Udall J A, Zhang X L. Reference genome sequences of two cultivated allotetraploid cottons,and.,2018, 51: 224–229.
[8] Du X, Huang G, He S, Yang Z, Sun G, Ma X, Li N, Zhang X, Sun J, Liu M, Jia Y, Pan Z, Gong W, Liu Z, Zhu H, Ma L, Liu F, Yang D, Wang F, Fan W, Gong Q, Peng Z, Wang L, Wang X, Xu S, Shang H, Lu C, Zheng H, Huang S, Lin T, Zhu Y, Li F. Resequencing of 243 diploid cotton accessions based on an updated a genome identifies the genetic basis of key agronomic traits.,2018, 50: 796–802.
[9] Zhang T, Hu Y, Jiang W, Fang L, Guan X, Chen J, Zhang J, Saski C A, Scheffler B E, Stelly D M, Hulse-Kemp A M, Wan Q, Liu B, Liu C, Wang S, Pan M, Wang Y, Wang D, Ye W, Chang L, Zhang W, Song Q, Kirkbride R C, Chen X, Dennis E, Llewellyn D J, Peterson D G, Thaxton P, Jones D C, Wang Q, Xu X, Zhang H, Wu H, Zhou L, Mei G, Chen S, Tian Y, Xiang D, Li X, Ding J, Zuo Q, Tao L, Liu Y, Li J, Lin Y, Hui Y, Cao Z, Cai C, Zhu X, Jiang Z, Zhou B, Guo W, Li R, Chen Z J. Sequencing of allotetraploid cotton (L. acc. TM-1) provides a resource for fiber improvement.,2015, 33: 531–537.
[10] Islam M S, Fang D D, Thyssen G N, Delhom C D, Liu Y L, Kim H J. Comparative fiber property and transcriptome analyses reveal key genes potentially related to high fiber strength in cotton (L.) line MD52ne.,2016, 16: 1–19.
[11] Hande A S, Katageri I S, Jadhav M P, Adiger S, Gamanagatti S, Padmalatha K V, Dhandapani G, Kanakachari M, Kumar P A, Reddy V S. Transcript profiling of genes expressed during fibre development in diploid cotton (L.).,2017, 18: 675.
[12] Trapnell C, Pachter L, Salzberg S L. TopHat: discovering splice junctions with RNA-Seq.,2009, 25: 1105–1111.
[13] Cole T, Adam R, Loyal G, Geo P, Daehwan K, Kelley D R, Harold P, Salzberg S L, Rinn J L, Lior P. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks.,2012, 7: 562–578.
[14] Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto encyclopedia of genes and genomes.,1999, 27: 29–34.
[15] Minoru K, Susumu G, Yoko S, Masayuki K, Miho F, Mao T. Data, information, knowledge and principle: back to metabolism in KEGG.,2014, 42: 199–205.
[16] Livak K J, Schmittgen T D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method.,2001, 25: 402–408.
[17] Wan Q, Guan X Y, Yang N N, Wu H T, Pan M Q. Small interfering RNAs from bidirectional transcripts ofregulate cotton fiber development., 2016, 210: 1298–1310.
[18] Wu H T, Tian Y, Wan Q, Fang L, Guan X Y, Chen J D. Genetics and evolution of MIXTA genes regulating cotton lint fiber development., 2018, 217: 883–895.
[19] Du S J, Dong C J, Zhang B, Lai T F, Du X M, Liu J Y. Comparative proteomic analysis reveals differentially expressed proteins correlated with fuzz fiber initiation in diploid cotton (L.).,2013, 82: 113–129.
[20] Daviere J M, Achard P. A pivotal role of DELLAs in regulating multiple hormone signals.,2016, 9: 10–20.
[21] Chen Z J, Guan X Y. Auxin boost for cotton.2011, 29.
[22] Xiao G H, He P, Zhao P, Liu H, Zhang L, Pang C, Yu J. Genome-wide identification of thegene family reveals thatandare involved in cotton fibre cell initiation.,2018, 69: 4323–4337.
[23] Han, X Cloning and expression analysis of novel Aux/IAA family genes in.,2012, 503: 83–91.
[24] Gilbert M K, Bland J M, Shockey J M, Cao H, Hinchliffe D J, Fang D D, Naoumkina M. A transcript profiling approach reveals an abscisic acid-specific glycosyltransferase (UGT73C14) induced in developing fiber of ligon lintless-2 mutant of cotton (L.).,2013, 8.
[25] Zhang D Y, Zhang T Z, Guo W Z. Effect of H2O2on fiber initiation using fiber retardation initiation mutants in cotton ().,2010, 167: 393–399.
[26] Taliercio E W, Boykin D. Analysis of gene expression in cotton fiber initials.,2007, 7: 22.
[27] Kudla J, Batistic O, Hashimoto K. Calcium signals: the lead currency of plant information processing.,2010, 22: 541–563.
[28] Steinhorst L, Mähs A, Ischebeck T, Zhang C X, Zhang X X, Arendt S, Schültke S, Heilmann I, Kudla J. Vacuolar CBL- CIPK12 Ca2+-sensor-kinase complexes are required for polarized pollen tube growth.,2015, 25: 1475–1482.
Analysis of differentially expressed genes and fiber development infuzzless mutant
WANG Xiao-Yang1, WANG Li-Yuan1, PAN Zhao-E1, HE Shou-Pu1, WANG Xiao1, GONG Wen-Fang1,3,*, and DU Xiong-Ming1,2,*
1Institute of Cotton Research, Chinese Academy of Agricultural Sciences / State Key Laboratory of Cotton Biology, Anyang 455000, Henan, China;2Zhengzhou Research Base, State Key Laboratory of Cotton Biology / Zhengzhou University, Zhengzhou 450001, Henan, China;3Key Laboratory of Cultivation and Protection for Non-wood Forest, Ministry of Education / Central South University of Forestry and Technology, Changsha 410004, Hunan, China
Cotton fiber, one of main nature textile and longest single cell, is a good material for researching the cotton fiber development. In this study, fuzz mutant (FZ) and wild-type () which belong to diploidspecies were used for unraveling molecular mechanism of fuzz fiber initiation by scanning electron microscopy (SEM), paraffin section and RNA sequencing technology. It was found that any fibers scarcely initiated on the ovule surface of the mutant at 0DPA, compared with that of the wild-type. Only lint fiber cells emerged on the ovule surface of fuzz mutant, while abundant fuzz and lint fiber cells appeared in wild-type at +3DPA stage. The transcriptome analysis of FZ andacross the four fiber development stages (0DPA, +3DPA, +5DPA, and +8DPA) indicated that the number of differentially expressed gene was 3780. There were litter differentially expressed genes at 0DPA in FZ and. In the proceed of ovule development, the number of differentially expressed genes was increased gradually. The KEGG enriched pathway analysis illustrated that these differentially expressed genes were involved in cutin, suberine and wax biosynthesis, phenylalanine metabolism pathways and plant hormone signal transduction pathways. The analysis of co-expressed gene trend lines indicated that the up-regulated genes in mutant at +3DPA were all involved in metal ion binding, MAPK cascade, oxidoreductase activity and transcription regulation, resulting in failed fuzz fiber cells initiation in mutant. These findings elaborated a dynamic variation about fuzz fiber initiation in diploid, which provides a reference for further research in the cotton fiber development.
;fuzzless mutant; differentially expressed gene; co-expressed trend
10.3724/SP.J.1006.2020.94133
本研究由国家自然科学基金项目(31601353)和国家重点研发计划项目(2017YFD0101601-03)资助。
This study was supported by the National Natural Science Foundation of China (31601353) and the National Key Technology R&D Program (2017YFD0101601-03).
杜雄明, E-mail: dxm630723@163.com; 龚文芳, E-mail: gwf018@126.com
E-mail: wangxiaoyang198806@126.com
2019-09-02;
2019-12-26;
2020-01-15.
URL: http://kns.cnki.net/kcms/detail/11.1809.S.20200115.1132.018.html
猜你喜欢
动物医学进展(2024年4期)2024-04-10 01:50:04
生物技术通报(2023年2期)2023-03-07 12:54:40
蔬菜(2022年7期)2022-07-29 15:47:37
纺织器材(2021年3期)2021-07-27 03:34:24
国际纺织导报(2021年8期)2021-03-17 09:56:56
纺织器材(2020年2期)2020-06-05 10:16:14
山东农业大学学报(自然科学版)(2020年2期)2020-05-21 00:18:06
心电与循环(2020年1期)2020-02-27 07:48:24
江苏农业学报(2015年6期)2015-03-26 10:57:58
湖北农业科学(2014年3期)2014-07-21 10:25:00
