
玉树牦牛与四个牦牛群体线粒体DNA D-Loop区遗传多样性分析
2020-04-16 02:14侯孟典宋仁德柴志欣杨玉文尕才仁汪永洲信金伟钟金城
家畜生态学报 2020年3期
侯孟典,宋仁德,王 会,柴志欣,杨玉文,尕才仁,汪永洲,信金伟,钟金城*
(1.西南民族大学 青藏高原动物遗传资源保护与利用教育部重点实验室,四川 成都 610041;2.青海省玉树州畜牧兽医工作站,青海 玉树 815099;3.青海省玉树曲麻莱县畜牧兽医工作站,青海 曲麻莱 815500;4.青海省玉树杂多县畜牧兽医工作站,青海 杂多 815300;5.西藏自治区农牧科学院省部共建青稞和牦牛种质资源与遗传改良国家重点实验室,西藏 拉萨 850009)
牦牛(Bos grunniens)是青藏高原特有的家畜品种,是我国重要的牛种资源,现已成为青藏高原地区畜牧业发展的独立板块[1]。玉树牦牛主要分布在海拔3 700 m以上的高寒地区,具有很强的适应能力[2]。经过长时间的自然选择,牦牛在形态、生理特性上已经获得稳定的遗传特征[3]。
线粒体DNA具有不易发生重组、母系遗传、变异速度快等特点,因此被广泛用于种群内或是种群间的遗传变异、遗传多样性、群体遗传结构分析等研究[4-6]。线粒体DNA D-环控制区(D-Loop),其进化速度比线粒体快10倍左右[7]。D-Loop区是线粒体DNA基因组中进化速率最快、最具遗传突变区的序列区域,由于自然选择等原因会存在巨大的变异,是目前常用的一种遗传标记方法[8-9]。
宋乔乔等[10]对西藏牦牛mtDNA D-Loop区序列多样性分析表明,西藏牦牛群体中的错那牦牛是比较纯的牦牛类群,日多牦牛、类乌齐牦牛、丁青牦牛、隆子牦牛、仲巴牦牛、聂荣牦牛、申扎牦牛等牦牛类群在进化过程中可能出现基因相互交流的情况;张成福等[11]研究发现,西藏牦牛可能存在两个母系起源;李瑞哲等[12]发现了青海雪多牦牛19种单倍型,可分为两个分支,推测雪多牦牛有两个母系起源;马志杰等[13]对野牦牛和中甸牦牛的D-Loop区分析,表明野牦牛具有丰富的遗传多样性;涂世英等[14]在中甸牦牛的研究中,以羊为外属对12个牛种进行对比分析,推测中甸牦牛可能与麦洼牦牛因地理位置相近而存在基因交流。
前人对于各个类群生物的mtDNA D-Loop区序列的遗传多样性都有相应的研究[15-25],但对玉树高原牦牛和类乌齐牦牛、斯布牦牛、申扎牦牛、帕里牦牛mtDNA D-Loop的比较研究还未见报道。本研究对玉树牦牛的遗传多样性进行探究,为玉树牦牛遗传资源的保护和利用研究奠定分子遗传学基础。
1 材料与方法
1.1 样品采集
共采集216头成年健康牦牛耳组织样品,其中青海省玉树州玉树牦牛64头、西藏自治区申扎县申扎牦牛36头、西藏自治区拉萨市斯布牦牛39头、西藏自治区日喀则亚东县帕里牦牛30头、西藏自治区类乌齐县类乌齐牦牛47头。组织于75%乙醇中保存带回实验室,放置在-80 ℃保存备用。
DNA提取试剂盒与DNA纯化回收试剂盒均购于天根生化科技有限公司(北京);DNA聚合酶及dNTPs购于Thermo Fisher Scientific公司(上海);2000 bp marker购于TaKaRa公司(大连)。
1.2 基因组DNA的提取及检测
使用DNA提取试剂盒(天根生化科技有限公司)提取各类群牦牛耳样的基因组DNA。提取产物用1%的琼脂糖凝胶电泳和紫外分光光度计检测其质量和浓度。置于-20 ℃冰箱保存备用。
1.3 引物设计及mtDNA D-Loop区序列的扩增
参照NCBI中已发布的牦牛mtDNA D-Loop区序列(AY:684273),使用Premier 5软件设计PCR扩增的上、下游引物。引物序列为F:5'CTACAGTCTCACCGTCAACC'3,R:5'GGGGTGTAGATGCTTGC'3,由上海赛默飞公司合成。
PCR扩增反应总体系25 L。其中灭菌ddH2O加入9.5 L、DNA模板加入1 L、上下游引物各加入1 L、2×long Taq聚合酶12. 5 μL。PCR反应程序:94 ℃预变性5 min;94 ℃变性30 s;58.5 ℃复性1 min;72 ℃延伸1 min;共35个循环;72 ℃ 终延伸5 min,16 ℃保存。扩增产物用1%的琼脂糖凝胶电泳检测其片段长度,粗略检测是否为目的条带。确认为目的条带的产物,经过胶回收试剂盒回收纯化后,由擎科新业生物技术有限公司(成都)测序。
1.4 数据处理
从NCBI中下载的牦牛mtDNA D-Loop区全序列,用BioEdit ClustalW Multiple alignment功能和DNAMAN对比分析D-Loop区与NCBI中发布的牦牛mtDNA D-Loop 区的差异,并辅以人工校对保证结果的可靠性及准确性。采用MEGA5.2软件统计mtDNA D-Loop区的碱基组成。使用DnaSPv5软件统计和计算序列中的单倍型数目(number of haplotypes,h)、单倍型多样度(haplotypediversity,Hd)、核苷酸多样性(nucleotide diversity,Pi)和平均核苷酸差异数(Average number of nucleotide differences,k)等群体遗传学指标,并做Tajima'sD中性显著性检验。运用MEGA 5.2软件中的邻接法(Neighbor-Joining,NJ) 功能对所有序列构建系统发育树。
2 结果与分析
2.1 5个群体牦牛mtDNA D-Loop区扩增结果分析
根据NCBI中牦牛mtDNA D-Loop区序列(AY:684273),设计PCR引物,经过PCR扩增后,扩增产物用1%琼脂糖凝胶电泳检,扩增目的片段大小约为1 000 bp(图1),与预期结果一致,表明扩增成功可进行下一步试验。
2.2 玉树牦牛与其余四个牦牛群体mtDNA D-Loop区序列遗传多样性分析
2.2.1 mtDNA D-Loop区核苷酸组成及变异 本实验对玉树牦牛和其他4个牦牛群体216个个体的D-Loop区序列进行分析。测得mtDNA D-Loop区长度为945 bp,胸腺嘧啶(T)、胞嘧啶(C)、腺嘌呤(A)、鸟嘌呤(G)四种核苷酸中A+T的平均比例为60.295% (60.163%~60.373%),C+G的平均含量为39.705%(39.627%~39.837%)。其中玉树牦牛四种核苷酸比例中平均A+T含量为60.372%, C+G含量为39.627%;类乌齐牦牛A+T含量为60.350%,C+G为39.650%;帕里牦牛A+T含量为60.267%,C+G含量为39.733%;申扎牦牛A+T含量为60.325%,C+G为39.675%;斯布牦牛A+T含量为60.163%,C+G为39.837%。表明5种牦牛mtDNA D-Loop区富含A、T碱基,具有一定的A/T碱基偏好性。
以牦牛线粒体D-loop(GenBank: AY374125.1)全序列为标准,分析216个个体D-Loop区,共发现序列中碱基的插入、缺失、颠换和转换4种变异位点共127处(图2)。玉树牦牛序列中共存在变异位点74个,占所有序列总变异位点的34%。所有变异位点主要集中在135~390 bp和695~820 bp处,位于序列的两端位置,在539~693 bp区间没有变异位点存在。排除所有变异位点中的36个插入和缺失位点,剩下转换和颠换位点共91个,这91个位点中转换位点有63个,主要存在于135~390 bp处;颠换位点28个,其中25个位点主要集中在755~850 bp区间;颠换位点中A↔T之间的转换15次,占颠换总数的53.57%;A↔C之间的转换7次,占25.00%;G↔C之间的转换5次,占17.86%;G↔T之间的转换1次,占3.57%。
2.2.2 mtDNA D-Loop区核苷酸单倍型及其多样性 使用DnaSPv5软件对5个牦牛群体216个个体的单倍型进行统计分析,共发现单倍型71个,单倍型多样度(Hd)为0.909±0.016,核苷酸多样性(pi)为0.082。各类群分别的单倍型数、单倍型多样度(Hd)、核苷酸多样性(Pi)、平均核苷酸差异数(K)见表1。
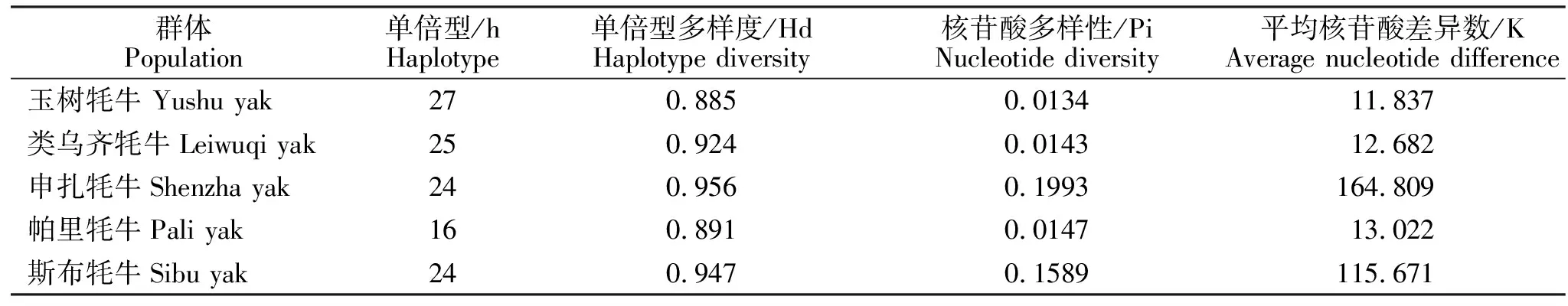
表1 5个牦牛群体的mtDNA D-Loop区序列单倍型数(h)、单倍型多样度(Hd)、核苷酸多样性(Pi)及平均核苷酸差异数(K)Table 1 mtDNA D-Loop region sequence haplotype, haplotype diversity, nucleotidediversity and average nucleotide difference in 5 yak populations
由表1可知,玉树牦牛具有最多的单倍型数。5个牦牛群体的单倍型多样度(Hd)在0.885~0.956之间,核苷酸多样性(Pi)在0.0134~0.1993之间,平均核苷酸差异数(K)在12.682~164.809之间。结果表明,各群体内个体间平均核苷酸差异数和核苷酸多样度呈正比例关系。各群体具有丰富的单倍型数目,玉树牦牛单倍型多样度、核苷酸多样性和平均核苷酸差异数均最低,申扎牦牛单倍型多样度、核苷酸多样性和平均核苷酸差异数均最高,说明其品种遗传一致性较高。
5个牦牛群体的Tajima's D检验见表2。表2表明玉树牦牛(P<0.05)Tajima's D检验差异显著,说明玉树牦牛不遵循中性进化,可能是自然选择导致群体扩增的结果。类乌齐牦牛、申扎牦牛、帕里牦牛和斯布牦牛Tajima's D检验(P>0.1)差异不显著,说明这4个牦牛群体符合中性进化。

表2 5个牦牛群体的Tajima's D检验Table 2 Tajima's D test of 5 yak populations
2.3 牦牛群体D-Loop区序列的遗传距离和系统进化分析
由表3可见,玉树牦牛与类乌齐牦牛和帕里牦牛的遗传距离较近(0.023、0.018),申扎牦牛与斯布牦牛的遗传距离最远(0.533)。
系统发育树也称为系统进化树,是一种用类似于树木分支样的图,用于表示物种之间的亲缘关系,进而可以用来推测物种的进化历史。核苷酸序列、蛋白序列等是构建系统发育树的主要依据[26]。利用MEGA 5. 2 软件中的邻接法( Neighbor-Joining,NJ) 对5个群体的216个个体的mtDNA D-Loop区序列构建系统发育树(图3),并做自举分析( Boot strap test) 1 000 次重复抽样检验系统树各分支的置信度。从图3中可以看出这216个个体主要有两个分支,斯布牦牛和申扎牦牛的14个个体为单独一支,其他个体为一支。

表3 5个牦牛群体mtDNA D-Loop区序列Kimura 双参数遗传距离Table 3 Kimura two-parameter genetic distance of mtDNA D-Loop region sequence of 5 yak populations
图4五个牦牛群体mtDNAD-Loop区序列NJ系统发育树
Fig.3 mtDNA D-Loop region sequence NJ phylogenetic tree
of five yak populations
3 讨 论
3.1 牦牛mtDNA D-Loop区序列核苷酸变异特征
本研究测定了216个牦牛个体的mtDNA D-Loop区序列,测得序列长度为945 bp,其中四种核苷酸A+T的含量为60.295% (60.163%~60.373%),C+G含量为39.705%(39.627%~39.837%),5个牦牛群体的核苷酸含量相差不大。王彦环等[27]、宋乔乔等[10]及张成福等[11]分别对夏南牛和西藏牦牛mtDNA D-Loop区中碱基含量进行分析,表明夏南牛与西藏牦牛A+T含量在60.9%~62%,与本研究的结果基本一致,均符合动物mtDNA中A/T含量偏高的特性。
对216个序列分析后共发现变异位点127个,占总分析位点的13.44%,玉树牦牛共发现变异位点74个,所有的变异位点都集中在序列的两端,因此推测两端序列为主要高变区域。颠换、转换、缺失、插入4种变异类型都存在,其中以转换为主与王彦环等[27]的研究结果相一致。变异位点中既有颠换又有转换的位点,颠换位点中A/T颠换占颠换总数的53.57%,与张成福等[11]对西藏牦牛的研究表明A/T颠换为主的结果基本一致,推测玉树牦牛mtDNA D-Loop区存在较多突变可能与该地区特殊的环境有关。
3.2 玉树牦牛与另外4个牦牛的遗传多样性
mtDNA具有高变的特征[28],D-Loop进化速度比线粒体快10倍左右,符合中性进化等优点,近年来广泛应用于动物遗传多样性的研究。分析结果表明216个个体共存在71个单倍型,其中玉树牦牛就存在27个单倍型,与涂正超等[29]用酶切的方法分析了5个牦牛群体90个个体的mtDNA D-Loop区表明牦牛mtDNA D-Loop区缺少多样性的结果存在差异,可能是由于试验方法及样本量不同所造成。
单倍型多样度(Hd)、核苷酸多样性(Pi)及平均核苷酸差异数(K)是衡量遗传多样性的重要指标。赖松家等[30]研究表明麦洼牦牛的平均核苷酸差异数最大(19),核苷酸多样度为0.02132,犏牛的平均核苷酸差异数最小为3.0,核苷酸多样度为0.00336。郭松长等[31]研究发现,青海环湖牦牛的单倍型多样性最高(0.9848) ,巴州牦牛最低(0.8000)。这些牦牛遗传多样性均高于本研究中牦牛群体,而本研究中玉树牦牛单倍型多样度、核苷酸多样性和平均核苷酸差异数均最低(Hd为0.885、Pi为0.0134、K为11.837),但是单倍型数最高,说明玉树牦牛群体可能经历过瓶颈效应,虽然生存在恶劣的生态环境中,但是由于人工选择等原因使遗传结构发生了较大改变。通过Tajina's D检验结果可知玉树牦牛中性检验显著,其余四个群体均为不显著,表明申扎牦牛、类乌齐牦牛、斯布牦牛和帕里牦牛符合中性进化,玉树牦牛可能由于自然选择等原因出现过群体扩张。
3.3 玉树牦牛与另外4个牦牛的遗传分化
根据Kimura 双参数模型计算群体遗传距离和NJ系统进化树分析可知,玉树牦牛、类乌齐牦牛、帕里牦牛、申扎牦牛和斯布牦牛可以分为两个大的分支。遗传距离分析结果可知玉树牦牛与申扎牦牛和斯布牦牛的遗传距离最远。玉树牦牛、类乌齐牦牛、帕里牦牛、申扎牦牛和斯布牦牛均属于西藏牦牛,13个申扎牦牛和斯布牦牛单独分为一支,其他个体和玉树牦牛分为一支,这与遗传距离分析的结果一致。说明申扎牦牛和斯布牦牛可能有两个母系遗传起源,与宋乔乔等[10]将西藏8个牦牛群体分为两大类的研究结果基本一致。
4 结 论
本研究用mtDNA D-Loop区分子标记方法分析玉树牦牛与4个西藏牦牛群体D-Loop区序列,表明这5个牦牛群体具有丰富的遗传性,玉树牦牛与类乌齐牦牛和帕里牦牛的遗传距离最近,与斯布牦牛和申扎牦牛的遗传距离相对较远。5个牦牛类群个体可以分为两个分支。
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
肝博士(2022年3期)2022-06-30
青海湖(2020年4期)2020-09-22
作文评点报·小学五、六年级(2020年24期)2020-06-21
故事作文·高年级(2020年3期)2020-03-17
小小说月刊(2019年20期)2019-10-19
好日子(2016年9期)2016-10-24
火花(2015年1期)2015-02-27
当代体育·扣篮(2014年11期)2014-07-08
