
尿素通道作为新型利尿药靶点的研究进展
2020-04-14 03:29张顺杨宝学
中国药理学与毒理学杂志 2020年12期
张顺,杨宝学
(北京大学基础医学院药理学系,天然药物及仿生药物国家重点实验室,北京 100191)
利尿药是临床上常用药物,主要用于治疗高血压、心衰和各种水肿性疾病等。其作用靶点在肾,通过增加尿量发挥利尿作用。但由于传统利尿药通过排钠排水利尿,会导致电解质平衡紊乱等副作用。科学家们一直在寻找新的利尿药靶点,研发新型利尿药。尿素通道(urea transports,UT)是一种新的利尿药靶点,其抑制剂有望发展成为不引起电解质平衡紊乱的新型利尿药。本文拟总结UT为利尿药靶点的理论基础、UT 抑制剂的筛选和评价方法及目前已被发现的小分子UT 抑制剂,为后续的以UT为靶点的新型利尿药研发提供参考。
1 尿素通道
UT 是一类选择性通透尿素的膜通道蛋白,在维持肾内尿素循环,建立肾髓质组织尿素浓度梯度和尿浓缩机制中发挥重要作用[1-5]。YOU 等[6]于1993 年首先克隆第一个UT-A2 的cDNA。迄今已克隆的UT 分为UT-A 和UT-B 2 个亚家族。UT-A亚家族由Slc14a2基因编码,包括6 个成员UTA1~UT-A6,其中UT-A1 和UT-A3 表达在内髓集合管末段的上皮细胞[7-8],UT-A2主要表达于髓袢降支细段[9],UT-A4在内髓集合管末段表达(只在大鼠发现)[7],UT-A5 表达于睾丸间充质细胞[10],UT-A6 表达于人结肠黏膜上皮细胞[11]。UT-B 由Slc14a1基因编码,广泛表达于全身多个组织器官,在肾直小血管内皮细胞、红细胞、脑、心、脾、结肠、睾丸和膀胱等组织表达水平较高[12]。
1.1 尿素通道的作用
UT 在维持肾内尿素循环和尿液浓缩中发挥重要作用。在肾集合管,精氨酸血管加压素通过调节水通道蛋白2上膜,增加水的重吸收,而尿素因不能透过集合管近端大部分节段而不断被浓缩,在内髓集合管末端达较高浓度。UT-A1 和UT-A3 表达于内髓集合管末端的上皮细胞,尿素通过UT-A1 和UT-A3进入内髓间充质组织,间充质组织高浓度尿素通过内皮细胞的微孔进入直小血管升支,随血液向肾皮质方向转运,在血管内外尿素浓度差的作用下,尿素从直小血管升支进入组织,通过UT-B进入直小血管降支,随血流再返回内髓,从而建立从肾外髓至内髓的尿素浓度梯度,以维持从肾皮质至肾髓质的渗透压梯度。因此,UT 在维持肾内尿素循环和尿液浓缩机制中发挥重要作用[13-14](图1)。
1.2 UT-B晶体结构的解析
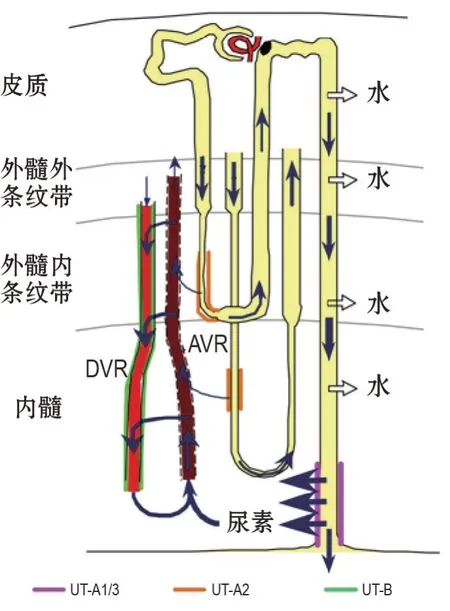
图1 肾内各尿素通道(UT)亚型的表达部位及肾内尿素循环示意图. DVR:直小血管降支;AVR:直小血管升支;➝:尿素移动方向.
UT-B 晶体结构的解析为基于结构寻找及优化UT抑制剂提供结构基础。UT家族成员的氨基酸序列具有相似的结构特征,通常包含10 个跨膜螺旋,其中UT-A1 包括20 个跨膜螺旋,由2 个UT结构域串联而成[15]。每个区域中前5 个跨膜螺旋与最后5 个跨膜螺旋具有明显的同源性[16]。而UT 另一个明显的结构特征是每5个跨膜螺旋都包含一个保守结构域,带有LPXXTXPF 序列,该序列在选择性通透尿素过程中发挥重要作用。研究表明,每个UT每秒可以通透1×104~1×106个尿素分子。UT-B 对甲酰胺和乙酰胺等小分子尿素类似物也具有选择性通透功能[17]。UT-B 的晶体结构已被解析,其以三聚体状态存在于细胞膜,每一个UT-B 分子结构相当于2个结构相似的半圆柱状结构域相向扣合而成的空心圆柱(图2A)。UT-B分子圆柱状结构的中心孔道可选择性通透尿素和乙酰胺等小分子尿素类似物。这一通道的两端分别称为So位点(向细胞外侧开口)和Si位点(向细胞内侧开口),孔道的中央称为Sm位点,位于So和Si之间;2个由3个氧原子组成的线性阵列的侧链被称为氧梯。在结晶过程中加入金原子能影响侧链在晶体结构中的位置[18](图2B、2C)。UT-B的三维结构使尿素分子部分脱水结合到孔道端口(So或Si位点),并可进一步彻底脱水移动到Sm位点,从而穿过孔道[19]。
2 尿素通道敲除小鼠模型
2.1 UT-B敲除小鼠模型
UT-B 功能性敲除导致尿素选择性尿浓缩能力下降。2002年,YANG等[12]通过敲除Slc14a1基因的3~6 号外显子建立了第一个UT-B 敲除小鼠模型。与野生型小鼠相比,UT-B 敲除小鼠的生长和发育无明显差异,但尿量增加50%,同时使尿渗透压降低25%[12]。UT-B敲除模型小鼠的肾内髓组织尿素浓度和渗透压降低,而钠、钾和氯等电解质浓度正常,且尿浓缩能力下降并不影响其他肾功能和机体的电解质平衡,提示UT 功能性缺失可提供阻断肾内尿素循环引起尿素选择性利尿。UT-B 全身敲除会导致小鼠血尿素浓度升高[12],尿素在海马蓄积可引起抑郁样行为[20-21]。UT-B 的敲除导致了尿素在睾丸的蓄积和雄性生殖系统早熟。此外,UT-B敲除可诱导膀胱尿路上皮细胞DNA 损伤和凋亡[22-23]。
2.2 UT-A敲除小鼠模型
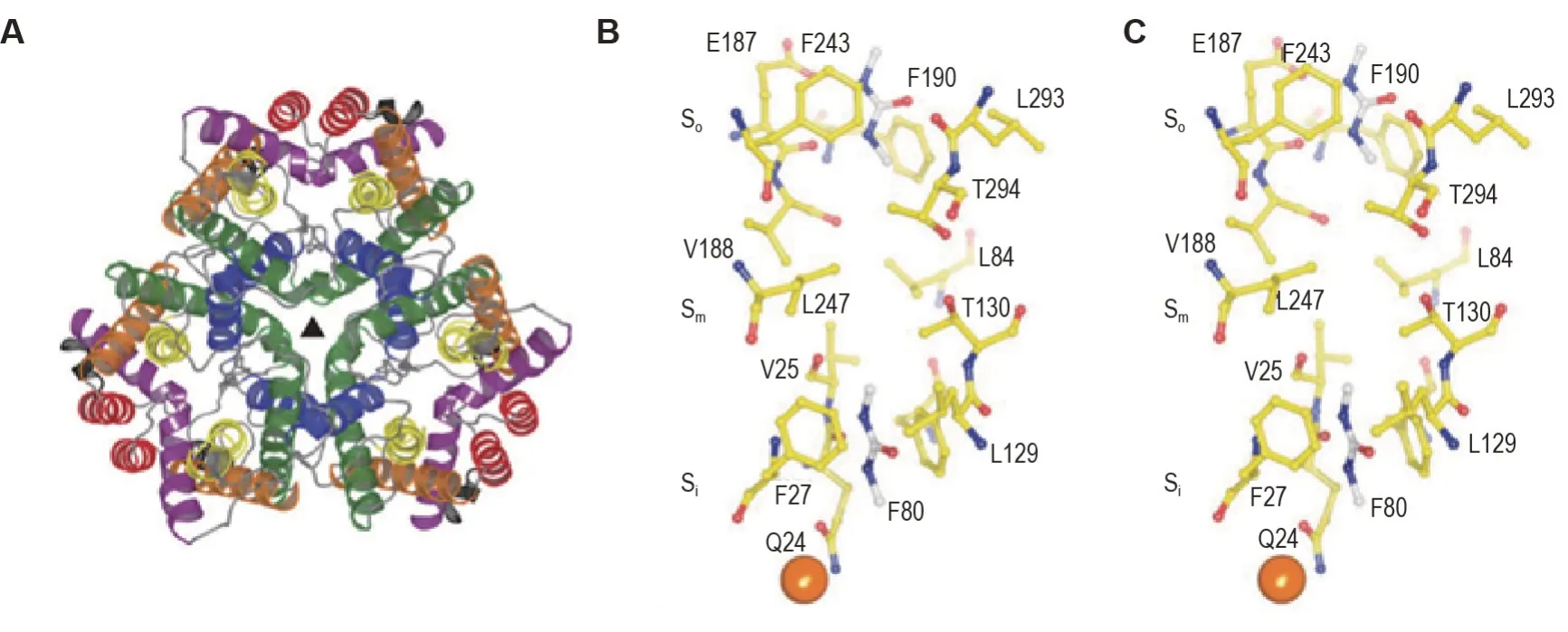
图2 脱硫弧菌尿素通道(dvUT)的晶体结构及氨基酸位点[18]. A:dvUT 三聚体结构,▲为晶体的3 次对称轴;B:在N,N′-二甲基脲(DMU)与dvUT结合位点,金原子与dvUT发生共结晶;C:金原子影响氧梯侧链在晶体结构中的位置.
UT-A1 在尿素选择性尿浓缩机制中发挥重要作用。敲除Slc14a2基因10 号外显子140 bp 大小的片段获得UT-A1/A3双敲除小鼠。与野生型小鼠相比,UT-A1/A3 敲除小鼠尿量增加2 倍,尿渗透压降低70%。在UT-A1/A3 敲除小鼠中,血浆尿素浓度与正常小鼠无明显差异,且除肾较小、睾丸较大外,未见其他明显异常[24-25]。敲除Slc14a2基因位于第12内含子的启动子及部分13号外显子的一个4 kb的基因片段,建立UT-A2敲除小鼠。在正常条件下或脱水条件下,UT-A2敲除小鼠与野生型小鼠相比,尿量及髓质尿素浓度无明显差异。而在低蛋白饮食(4%蛋白质)的情况下,UT-A2 敲除小鼠最大尿渗透压及内髓尿素浓度显著降低[26]。在UT-A1/A3 双敲除小鼠转入UT-A1 基因,获得了肾集合管末段只缺失UT-A3基因的小鼠,该小鼠的基础尿素通透性及尿浓缩能力达到正常水平,提示UT-A1在肾集合管末段的重要性远大于UT-A3[27]。对UT-A1编码基因Slc14a2的9号外显子后的“gt”拼接信号进行同义突变,在不影响其他UT-A 成员转录和翻译的同时,获得UT-A1 特异性敲除小鼠。UT-A1敲除小鼠的尿量约为野生型小鼠尿量的3倍,其内髓组织液尿素浓度明显低于野生型小鼠,而Na+,K+和Cl-浓度与野生型小鼠相比无明显差异。UT-A1敲除小鼠的生长发育、肾功能指标、血电解质水平及肾组织结构方面均无明显异常[28]。
2.3 全部尿素通道敲除小鼠模型
全部UT 敲除只降低尿浓缩能力,无明显其他表型异常。JIANG 等[29]通过同时敲除Slc14a1和Slc14a2基因的大部分片段,成功构建了全部UT敲除小鼠模型。与野生型小鼠相比,模型小鼠日尿量增加近3.5 倍,尿渗透降低约75%。由于敲除了所有UT,尿素在内髓集合管末端进入内髓组织和通过直小血管进入内髓组织减少,髓组织尿素浓度显著低于野生型小鼠,而内髓Na+,K+和Cl-浓度与野生型小鼠无明显差异,证实UT 功能性敲除通过破坏肾内尿素循环,减小内髓组织尿素产生的渗透压梯度,导致尿浓缩能力降低。
2.4 不同尿素通道敲除小鼠模型尿指标比较
UT 亚型功能缺失小鼠的相对尿量比较结果显示,全部UT 敲除小鼠>UT-A1/A3 双敲除小鼠≥UT-A1 敲除小鼠>UT-B 敲除小鼠>UT-A3 缺失小鼠>UT-A2 敲除小鼠≥野生型小鼠;相对尿渗透压排序为:UT-A1/A3 双敲除小鼠≤全部UT 敲除小鼠 由于红细胞较易获得且天然高表达水通道蛋白1(aquaporin-1,AQP1)和UT-B,红细胞裂解筛选模型适用于高通量筛选UT 抑制剂[31]。2007 年,VERKMAN 团队[31]开发了一种应用红细胞裂解高通量筛选UT-B 抑制剂的实验模型,该模型的原理基于红细胞膜上AQP1 对水的通透及UT-B对尿素的通透(图3A)。该筛选模型操作简单、方便。首先将红细胞置于含2 mol·L-1尿素或尿素类似物乙酰胺的磷酸盐缓冲溶液中孵育2 h,使红细胞内尿素浓度达到2 mol·L-1,再将红细胞迅速置于等渗磷酸盐缓冲溶液中。由于红细胞内外的渗透压差的作用,水通过AQP1 快速进入红细胞,使红细胞体积膨胀。而尿素则在细胞内外尿素浓度差的作用下,快速通过UT-B 转移至细胞外,使红细胞内外的渗透压迅速到达平衡,红细胞体积恢复正常。如有UT 抑制剂存在,其将抑制尿素通过UT-B出细胞,细胞内外保持高渗透压差,促使水持续通过AQP1进入细胞,最终导致红细胞胀破。通过检测红细胞破裂所产生的血红蛋白的量,可发现对UT 有抑制作用的线索化合物。 停留实验(stopped-flow)可用于研究溶液体系的毫秒级动力学过程,是验证UT 抑制剂特异性和有效性的常用方法(图3B)。将红细胞悬液与高浓度尿素溶液在stopped-flow仪器混匀小室中快速接触。细胞外高浓度尿素形成的高渗透压促使细胞内水通过AQP1快速流出,导致红细胞皱缩,随后尿素在细胞内外浓度差的作用下进入细胞,使细胞内外的渗透压差发生逆转,导致水回流入红细胞中,使细胞膨胀。红细胞的体积变化会影响穿过混匀小室的散射光的量,测量530 nm处90°散射光强度可定量分析细胞的体积变化,进而计算红细胞细胞膜对尿素的通透性[32]。 UT-A1 较UT-B 更适合作为利尿药靶点,因此需要一种能高通量筛选UT-A1 抑制剂的细胞模型。FROHLICH 等[33]将UT-A1 基因克隆到pcDNA5/FRT 中,并转染到具有完整翻转酶(flippase,Flp)重组靶位点的Madin-Darby 犬肾细胞MDCK中。毛喉素和精氨酸血管加压素可增加UT-A1的磷酸化,促进其从细胞内囊泡转移至质膜,介导尿素的跨膜通透。利用14C标记的尿素分子观察稳定表达UT-A1的MDCK细胞对尿素的通透速率,可检测UT抑制剂对UT-A1的抑制作用。基于类似的原理,将黄色荧光蛋白YFP-H148Q/V163S,AQP1和UT-A1(或UT-B)基因稳定转染入MDCK 细胞系,将MDCK细胞系培养于黑壁96孔培养板,对Cl-敏感的YFP-H148Q/V163S 能指示细胞体积的变化。减小细胞体积可使细胞内Cl-浓度增加,从而导致YFP荧光降低(图3C)。将细胞置于尿素溶液中,由于细胞外尿素浓度引起的渗透压差变化会驱使水外流和细胞的皱缩,进而尿素和水进入细胞恢复细胞的正常体积。可通过检测不同浓度的尿素浓度梯度下荧光信号强度变化,计算细胞体积的改变速率,分析细胞膜对尿素通透性,筛选和评价UT-A1抑制剂的作用[34]。 适合临床的新型利尿药还需进一步研究。1970 年,MACEY 和FARMER[35]研究发现,根皮素可在不显著改变水渗透系数的情况下,抑制尿素的渗透系数。硫脲类等尿素类似物在50~100 mmol·L-1的浓度下可有效改变红细胞对尿素的通透速率,也可抑制离体内髓集合管对尿素的通透[31,36]。但这些UT 抑制剂的半数有效剂量过大,缺乏成药性。2012 年,CIL 等[37]发现,使用尿素类似物二甲基硫脲(图4A)对UT-A1和UT-B抑制作用的IC50值分别为6.6和3.9 mmol·L-1,可显著增加尿量,降低尿渗透压。二甲基硫脲作用迅速、持续和可逆,对低蛋白饮食的大鼠利尿作用较差,且对血浆Na+,K+和Cl-的水平无明显影响,提示二甲基硫脲可用于确认UT作为利尿药靶点,但有效剂量太高,不适于新型利尿药的开发[31]。随后,ESTEVA-FONT等[38]测量了36个硫脲类似物对UT-A1和UT-B抑制活性,发现3-硝基苯基-硫脲(图4B)对UT-A1 和UT-B 的抑制作用的IC50均为0.2 mmol·L-1;而4-硝基-苯基硫脲是一种选择性UT-A1 抑制剂,对UT-A1 和UT-B 抑制作用的IC50分别为1.3 和10 mmol·L-1。然而,以上化合物的药理学特性较差,在临床上不适合研发成为新型利尿药。 图3 尿素通道抑制剂的筛选和药效学评价实验模型.A:红细胞裂解模型;B:停留实验(stopped-flow)模型;红细胞悬液溶于等渗PBS溶液,而高浓度尿素溶于等渗PBS溶液制成高渗溶液;C:荧光指示方法筛选模型;RBC:红细胞;AQP1:水通道蛋白1;YFP:黄色荧光蛋白;→:移动方向. 图4 已发现的尿素通道抑制剂的化学结构. UT-B 小分子抑制剂还需进一步改进。迄今,通过对不同小分子化合物库的高通量筛选,已经发现了多种具有明显UT-B 抑制活性的小分子化合物[34]。利用红细胞裂解筛选模型,VERKMAN 团队[31]发现了近30种小分子UT-B抑制剂,主要包括苯磺酰唑类(图4C)、苯磺酰胺类(图4D)、氨基酞嗪类(图4E)和氨基苯并咪唑类活性化合物(图4F)。具有苯磺酰胺结构的化合物(ureainh)-201 对人UT-B 的抑制活性达微摩尔以下水平(IC50=0.3 μmol·L-1),但对小鼠和大鼠UT-B 的抑制作用弱,药物浓度25 μmol·L-1仍未显示出抑制作用,限制了其进一步的动物实验研究[31]。氨基酞嗪类化合物PU1424,对人和小鼠UT-B 的IC50值分别为0.02和0.69 μmol·L-1,该化合物抑制作用部位在UT-B的So位点,对UT 通透尿素的抑制作用是可逆的[39]。但这些化合物在体内均无利尿活性。随后,YAO等[40]通过红细胞裂解模型从10万个化合物中发现三唑并噻吩并嘧啶类化合物具有明显的UT-B抑制活性,其中抑制率最高的化合物为UTBinh-14(图4G),该化合物对人UT-B和大鼠UT-B的IC50分别为10 和25 nmol·L-1,可完全可逆地抑制尿素的转运。UTBinh-14通过作用于UT-B 通道的细胞内位点,对尿素的转运产生竞争性抑制作用。同时研究发现,UTBinh-14 对UT-B 具有高度选择性,即使在较高浓度下,对UT-A1也无抑制作用。UTBinh-14由于其在肝微粒体中的快速代谢且对UT-A1 无活性而不适合作为临床应用的利尿药[34,41]。 已发现的UT-A1抑制剂虽具有体外抑制活性,但多数在体内无明显的利尿作用。ESTEVAFONT 等[34]使用YFP,UT-A1 和AQP1 共转染的MDCK细胞高通量筛选UT-A1小分子抑制剂,发现了具有UT-A1选择性或UT-A1/UT-B非选择性抑制活性的小分子,包括8-羟基喹啉类(图4H)、氨基噻唑啉类(图4I)和[1,3,5]三嗪类(图4J),其非竞争性地抑制对尿素通透。同时,它们对竞争性抑制尿素转运功能的三唑并噻吩并嘧啶类化合物进行结构优化后得到化合物UTA1inh-D1,对UT-A1有较高的选择性,抑制UT-A1 和UT-B 的IC50值分别为3.8和15 μmol·L-1。同时,他们的研究结果表明,8-羟基喹啉类、氨基噻唑啉类抑制剂的结合位点位于细胞内通透尿素的孔隙附近,而[1,3,5]-三嗪类药物则位于细胞外通透尿素的孔隙附近。LEE 等[42]发现的2,7-双取代芴酮(图4K)是一种新型的UT-A和UT-B抑制剂,它们与UT-A1在胞质的区域结合,可非竞争性地、可逆地抑制UT-A1活性。上述抑制剂虽具有体外抑制活性,但在体内均无明显的利尿作用。2018年,LEE等[43]通过高通量筛选发现了强效的UT-A1 选择性抑制剂1,2,4-三唑喹喔啉类化合物,其对UT-A1具有强效的抑制作用。其中活性最强的化合物8ay通过芳基醚连接1,2,4-三唑[4,3-α]喹喔啉与苯磺酰胺,以一种非竞争性机制快速、可逆地抑制UT-A1对尿素的通透,其对UT-A1的IC50约为150 nmol·L-1,对UT-B的IC50约为2 μmol·L-1。在8ay体外代谢实验中发现,喹喔啉环氧化后产生一种代谢稳定的7,8-二氟喹喔啉类似物8bl(图4L),静脉注射8bl 4 mg·kg-1可使小鼠尿量增加,尿渗透压降低。 UT-A1和UT-B共同小分子抑制剂的口服生物利用度有待提高。2013年,杨宝学课题组[32]通过高通量筛选发现,噻吩并喹啉类化合物PU21对UT-B和UT-A1 都有抑制作用。计算机分子对接分析提示,PU21 与UT-B 分子的ASN289 形成氢键,与LEU285 和ALA327 形成很强的范德华力和疏水作用,分子动力学模拟预测PU21 以铆钉方式结合在UT-B 的功能对接基团,抑制UT-B 通透尿素,从而发挥其对UT-B的抑制作用[44]。LI等[45]对噻吩并喹啉类化合物的类似物进行筛选,并经结构优化后得到活性化合物PU14(图4M),其对人、大鼠和小鼠的UT-B具有明显的抑制活性。PU14对UT-B敲除小鼠红细胞的尿素通透性无影响,说明PU14 对UT-B 介导的尿素通透性具有特异性的抑制作用。体内药效学结果表明,PU14 可剂量依赖性地增加大鼠尿量,降低尿渗透压。PU14 可降低大鼠肾内髓渗透压及尿素浓度,而不影响非尿素溶质的浓度,同时PU14 不影响血Na+,K+和Cl-的水平,说明PU14 在发挥利尿作用的同时不影响电解质平衡[45]。REN 等[46]在对PU14 结构优化后,得到对UT-B 和UT-A1 抑制活性更强的化合物PU48(图4N)。大鼠皮下注射PU48 后,出现剂量依赖性地尿量增多,PU48在大鼠体内快速吸收和消除,并迅速到达其消化道、肝、肾和膀胱等靶器官[47-48]。但因其溶解度差、生物利用度低和无口服利尿活性等限制了PU48的进一步开发。通过对噻吩并喹啉的结构修饰,ZHAO等[49]和LI等[50]获得了新型特异性的UT 抑制剂噻吩并吡啶类化合物。通过结构优化,得到化合物8NP(图4O)和CB-20(图4P),其对UT-A1和UT-B 的抑制活性几乎相同,且对大鼠有良好的利尿活性,其水溶性明显优于噻吩并喹啉类UT 抑制剂。给动物皮下注射后,CB-20 产生了良好的利尿活性,血中Na+,K+和Cl-未发生明显的改变。长期服用CB-20 可持续稳定地增加尿量。同时,CB-20在大鼠体内吸收良好且清除速率较快。但也因其口服生物利用度过低,不宜于进行药物开发。 口服给药是一种比较适于临床长期治疗的给药方式,其具有给药方式简便快捷,由胃肠道吸收,不引起皮肤或黏膜损伤等优点。为获得具有口服活性的UT抑制剂,ZHANG等[51]根据药物化学的原理筛选了1040 种具有疏水性结构的小分子尿素类似物,并发现具有二芳基酰胺母核的新型UT 抑制剂,其中化合物1H 是目前发现的第一个具有口服利尿作用的UT 抑制剂。灌胃给予100 mg·kg-1的1H 后,大鼠尿量明显增加而尿渗透压明显降低,给药后第0~2 小时出现显著性差异,给药后第2~4 小时差异最明显,利尿效果可持续6 h,而非尿素溶质的排泄量无明显变化。而连续给药的结果显示,1H可持续性地增加大鼠尿量、降低尿渗透压,不影响非尿素溶质的排泄量。1H连续给药后,大鼠肾内髓组织渗透压和尿素浓度明显降低,而非尿素溶质的浓度无明显变化,说明1H 主要是通过影响内髓组织尿素浓度影响肾内髓组织渗透压梯度。1H 对大鼠UT-A1的IC50(0.097 μmol·L-1)明显低于对UT-B的IC50(0.64 μmol·L-1),是UT-A1高选择性的UT抑制剂。分别灌胃给予UT-A1敲除小鼠和UT-B 敲除小鼠100 mg·kg-1的1H,发现UT-B敲除小鼠在给药后尿量明显增加,而UT-A1敲除小鼠的尿量无显著变化,验证了1H对UT-A1的选择性。使用Caco-2细胞模型发现,1H具有良好的膜通透性,在人胃肠道中具有良好的吸收效果。大鼠体内药动学结果显示,1H在大鼠胃肠中吸收良好,血药浓度迅速达到有效水平。血清学指标检测发现,1H不导致传统利尿药引起的低钾血症、低钠血症和高尿酸血症等不良反应,也不影响正常肾功能及糖代谢、脂质代谢等生理学过程。然而,1H的口服生物利用度为4.38%,需要通过改进剂型、结构优化等方式进一步改善其成药性。 综上所述,由于目前临床上常用的利尿药普遍存在电解质失衡等不良反应,研究人员正试图以UT 为新的利尿药靶点研发新型利尿药。然而目前尚无UT 抑制剂在临床上用于患者的治疗。对UT抑制剂的研发存在一定的困难:由于UT-B 和UT-A1 结构具有相似性,难以发现高选择性抑制UT-A1 的UT 抑制剂;UT-A1 的晶体结构尚未被解析,阻碍了基于结构的UT 抑制剂的研发;肝硬化、心衰等低钠性水肿疾病模型的造模方法稳定性较差,阻碍了UT抑制剂在疾病模型中的研究;寻找一种口服活性较好且生物利用度高的UT抑制剂是新型利尿药研发的瓶颈。具有口服利尿活性的化合物1H 的发现为UT 抑制剂在临床的应用带来了希望,然而1H仍存在血浆稳定性差等缺点,尚需对其进行进一步的结构优化。随着对UT-A1 结构解析以及基于结构的药物筛选方法、化合物结构优化技术等不断成熟,加之药理学、药物化学、药动学和药剂学等学科的不断发展,必将会成功研发出以UT为靶点的新型利尿药,其可用于治疗充血性心衰、肝硬化腹水、肾病综合征等低钠性水肿相关疾病。3 尿素通道抑制剂的筛选方法
3.1 红细胞裂解筛选模型
3.2 停留实验
3.3 细胞荧光指示法筛选模型
4 已发现的尿素通道抑制剂
4.1 尿素类似物
4.2 UT-B小分子抑制剂
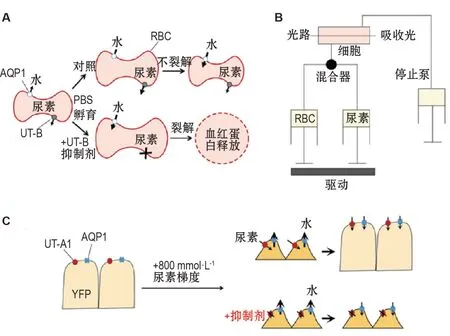
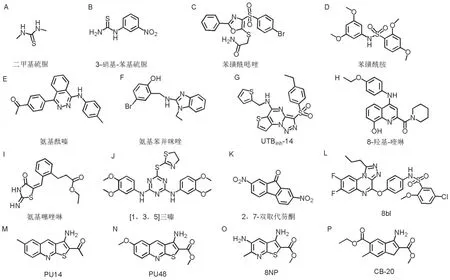
4.3 UT-A1小分子抑制剂
4.4 UT-A1和UT-B共同小分子抑制剂
4.5 口服尿素通道小分子抑制剂
5 结语
猜你喜欢
中国化肥信息(2022年9期)2022-11-23
求学·理科版(2022年4期)2022-04-03
世界科学技术-中医药现代化(2021年9期)2021-12-31
中国畜禽种业(2021年7期)2021-12-03
今日农业(2020年13期)2020-12-15
今日农业(2020年13期)2020-08-24
中西医结合心血管病杂志(电子版)(2020年13期)2020-06-23
福建基础教育研究(2020年2期)2020-05-28
今日农业(2019年12期)2019-08-13
安徽医学(2018年3期)2018-04-19
