
染色体相互作用密度与拓扑域相关分析
2020-04-08 09:30许希伦
电脑知识与技术 2020年3期
许希伦
摘要:生物信息学是一门交叉科学,利用计算机方法来揭示大量复杂生物数据所包含的生物学意义。染色体相关结构域是染色体上相互作用密集的一种重要结构,但目前缺少相互作用与拓扑域相关性分析。针对此问题,使用计算机分析方法,设计了相互作用密度指标,可以较好地表示相互作用地聚集程度,并分析了相互作用密度和拓扑域的特点和相关性。
关键词:生物信息学;染色体相互作用;染色体拓扑结构域;相互作用密度;相关性分析
中图分类号:TP3-05 文献标识码:A
文章编号:1009-3044(2020)03-0011-02
1 概述
在过去,人们主要依赖显微技术来研究染色体的空间组织模式。受显微技术的限制,观察的精度有限。随着染色质构象捕获技术的发展,可以通过Hi-C实验[1]测序得到染色体在三维空间中相互接近的片段。通过对Hi-C数据的分析,可以得到染色体上两个区域间的相互作用强度,并计算得到一种染色体结构——拓扑相关结构域,简称拓扑域[2-4]。拓扑域是染色体中重要的结构单元,它广泛存在于多种物种间,是一种保守的结构。这种结构在同一生物的不同细胞系间十分稳定,具有很高的相似性。对拓扑域的研究将有助于我们了解染色体发挥功能的机制,同时也有助于揭露染色体结构相关疾病的发生原因,为治疗相关疾病提供帮助[5-7]。
本文中我们将分析拓扑域与相互作用的性质,并提出相互作用密度的指标,来表示拓扑域中相互作用的密集程度。
2 实验分析
本文中,我们将分析相互作用的性质,拓扑域的性质和相互作用密度与拓扑域的相关程度。我们使用两种常用的细胞系IMR90(人胚肺成纤维细胞),并使用DI算法和ICFinder算法得到的拓扑域来进行分析。
为了构建染色体上的相互作用矩阵,我们首先将染色体按照40Kb的窗口大小分割成一个个等长且连续的区间,可依次记为B1,B2,…,Bn(假设分成n个区间)。之后根据Hi-C数据构建的相互作用矩阵定义为Mnxn其中第i行,第j列元素Mi,j表示区间Bi与Bj的相互作用强度。
2.1 相互作用分析
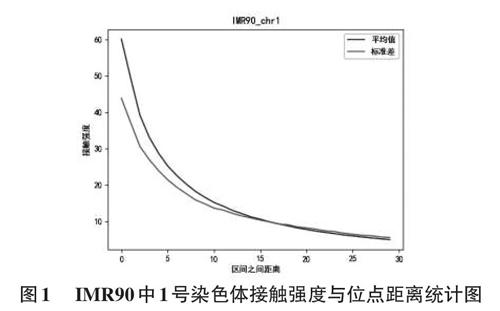
本小节我们比较了IMR90细胞中相互作用强度与距离的关系,其结果如图1所示。其横坐标表示位点之间的距离l,实线表示两个位点在当前距离下相互作用的平均值,距离以40kb为一个单位。虚线表示其标准差。
可以看到,随着两个位点距离的增加,之间的相互作用强度下降明显。并且其方差在不同距离下都较大,说明即使两个位点距离相同,其相互作用强度也有很大差异。
由此我们可以得出结论,两个位点之间的相互作用受距离影响明显,随着两个位点距离变远,其相互作用的强度会下降。
2.2 染色体拓扑域分析
在本节中,我们采用两个经典DI算法[4]和ICFinder算法[5]得到的拓扑域,来探究拓扑域的性质。本节我们使用IMR90细胞系的1号染色体为例,使用两种不同的算法,得到其拓扑域检测结果,并进行对比。
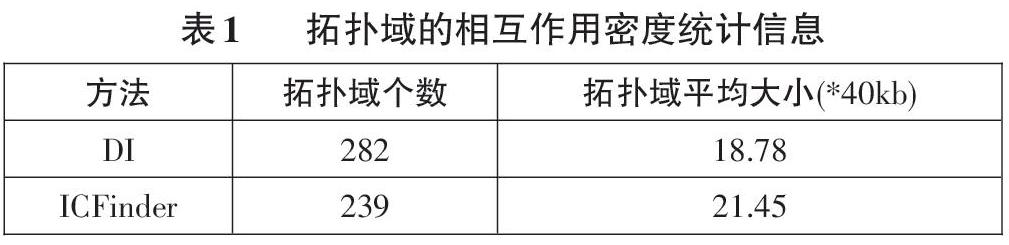
表1统计了两个检测结果的信息,可以看到,两个结果间的拓扑域平均大小均在20个单位。
2.3 相互作用密度与拓扑域分析
染色体拓扑域是染色体上相互作用密集的区域,也即在染色体上的两个位置间的相互作用相较于域外或者跨域邊界这两种情况,在拓扑域内其相互作用会明显较大。为了让不同距离的两个位置间的相互作用具有可比性,并且考虑其全局信息,我们对每个相互作用的两个位置的距离,根据前面介绍的平均值,对其转换得到一个规范化的相互作用矩阵Ⅳ。
如果Ni,j大于1,说明这两个位置间的相互作用大于全局的均值。根据规范化后的矩阵Ⅳ,我们提出了相互作用密度的概念。相互作用密度主要用于描述一个区域内部相互作用相较于全局的密集程度,值越大,说明其密集程度越大。
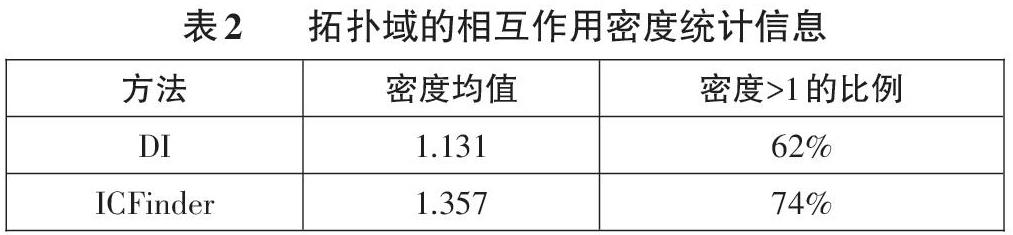
为了验证其有效性,我们使用IMR90细胞系的1号染色体,使用DI和ICFinder算法得到的拓扑域,我们统计了其结果的基本信息,如表2所示。可以看到,拓扑域的密度均值大于1,且ICFinder拓扑域中密度大于1的拓扑域比例达到74%。
我们计算其拓扑域的相互作用密度的分布,得到的分布图如图2所示。可以看到,其大部分拓扑域的密度均集中在大于1的部分。由此我们可以得出结论,我们所定义的相互作用密度可以有效地体现拓扑域内相互作用的密集程度,密度越大,表示其密集程度越高。
接下来我们继续探究拓扑域长度对相互作用密度的影响,我们统计了两个方法得到的拓扑域中,不同长度拓扑域的平均密度。其结果如图3所示,随着拓扑域长度的增大,其相互作用密度也随之增大。由此可以得出结论,在拓扑域内,长间隔的两个区间的相互作用强度较短间隔相比,其与全局平均水平差异更大。
3 结论
在本文中,我们分析了相互作用矩阵和已有算法检测得到的拓扑域的特点,并基于我们的发现,提出了相互作用密度的指标,该指标考虑到了距离的因素,可用于描述染色体上相互作用的聚集程度。通过分析该指标与已有算法检测得到的拓扑域的关系,我们发现该指标有以下特点:1)已有算法检测的拓扑域其相互作用密度大部分均大于1;2)随着拓扑域的增大,其相互作用密度也随之增大。
参考文献:
[1] Lieberman-Aiden E,Van Berkum N L,Williams L,et al.Comprehensive mapping of long-range interactions revealsfolding principles of the human genome[J]. science, 2009, 326(5950):289-293.
[2]张文力,高通量测序数据分析现状与挑战[J].集成技术,2012 (3):20-24.
[3]吕红强,郝乐乐,刘源,等,基于生物信息学的Hi-C研究现状与发展趋势(三维基因组专刊稿件)[J].遗传,2019: 0-0.
[4] Dixon J R,Selvaraj S,Yue F,et al.Topological domains inmammalian genomes identified by analysis of chromatin inter-actions[J]. Nature, 2012, 485(7398):376.
[5] De Laat W. Duboule D.Topology of mammalian developmen-tal enhancers and their regulatory landscapes[J]. Nature. 2013.502(7472):499-506.
[6] Pombo A,Dillon N.Three-dimensional genome architecture:players and mechanisms[J]. Nature reviews Molecular cell biol-ogy, 2015, 16(4):245-257.
[7] Matharu N,Ahituv N.Minor loops in major folds: enhancerproruoter looping, chroruatin restructuring, and their associa-tion with transcriptional regulation and disease[J]. PLoS genet-ics, 2015, 11(12):e1005640.
[8] Haddad N,Vaillant C,Jost D.IC-Finder: inferring; robustlythe hierarchical organization of chromatin folding[J]. Nucleicacids research. 2017, 45(10):81.
猜你喜欢
