
多酸在电催化析氢反应中的应用研究进展
2020-04-02 02:52葛靖暄胡钧朱盈婷泽妮诗臧德进秦召贤黄毅超张江威魏永革
物理化学学报 2020年1期
葛靖暄,胡钧,朱盈婷,泽妮诗,臧德进,秦召贤,黄毅超,*,张江威,*,魏永革,*
1有机光电子与分子工程教育部重点实验室,清华大学化学系,北京 100084
2催化基础国家重点实验室,中国科学院大连化学物理研究所,辽宁 大连 116023
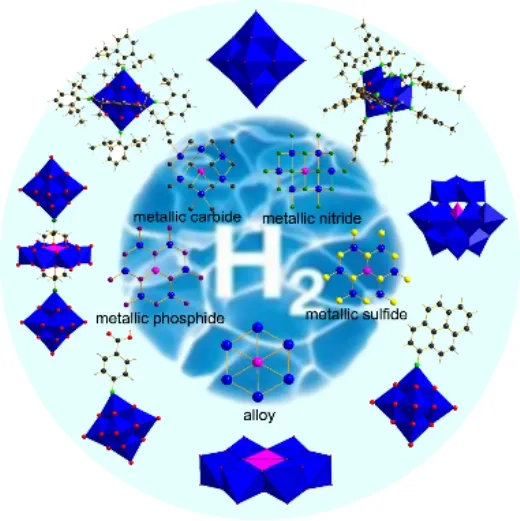
1 引言
随着全球气候变暖以及化石能源快速消耗,人们逐渐将目光转向可持续能源体系1-5。氢气是一种清洁、高效、无碳排放的能源载体,备受青睐。氢能也被认为是一种非常具有前景的化石燃料替代品6,7。特别是近年来,基于氢-氧燃料电池的新能源汽车逐渐成为一项未来绿色交通可行的方案。丰田公司已经率先向市场投入了第一代氢能源汽车,近日更是公布了数千项相关专利,预计2020年将售出30000辆燃料电池汽车(fuel cell vehicles,FCV)8。随着氢燃料电池的进一步普及和新时代环保要求的提高,氢气的低成本、清洁高效生产将是未来发展的必然趋势。
据统计,绝大多数的氢气是通过甲烷蒸汽重整和煤气化获得的(约95%),仅有少量是通过电催化水裂解(约4%)获得的9。然而前两种制氢的方法需要巨大的能耗和产生大量碳排放,加重温室效应10,11。因此,电解水制氢被认为是最有希望的绿色产氢途径之一12-18,但是由于析氢过电位的存在,过程中不可避免地会产生大量的能源消耗。因铂具有较高的电催化活性,常被用作商用阴极析氢电极材料,然而,铂价格昂贵,导致成本过高,从而限制了其工业化生产。
电解水包括氢气析出反应(Hydrogen Evolution Reaction,HER)以及氧气析出反应(Oxygen Evolution Reaction,OER)两个反应过程。开发廉价、高效、稳定的电催化剂是电解水制氢走向更广泛工业化应用的重要保障,也是广大科研工作者的需要攻克的目标12,13,19-24。近年来,科研工作者在电催化水裂解方面取得了长足进展。
很多高效的HER催化剂(包括:合金材料19,23,25,26、过渡金属硼化物27-29、碳化物30-36、氮化物20,34,37-40、氧化物3,41-44、磷化物30,32,35,45-56、硫化物14,17,21,22,30,52,57-79以及单原子催化剂12,80,81等)被开发出来。虽然大量的HER电催化剂已被报道,但是,这些催化剂仍存在活性和稳定性较差的问题,与商业Pt/C催化剂仍有一定差距。
多金属氧酸盐(简称:多酸)是由钒、钼、钨等过渡金属原子和氧配体结合形成的一类具有纳米尺寸的多金属阴离子氧簇合物82-86。多酸结构丰富,而且具有独特的光电磁特性,在医药87-90、催化83,91-95、材料96-98和能源99-104领域均具有广泛的应用。近几年多酸用于电催化水裂解方面的工作越来越多,这是因为一方面,多酸结构确定,又是单分散的纳米团簇,有利于暴露出更多的催化活性位点;另一方面,通过设计不同过渡金属取代的多酸,除了可以很好地提供钒、钼和钨源以外,还能提供原子精确的其他过渡金属原子。此外,通过有机修饰或者主客体结合形成多酸有机无机杂化材料,经处理后有望获得可控原子掺杂的超细纳米材料21,32,58,105,106。虽然一些综述报道的水裂解工作涵盖了部分多酸13-16,36,38,107-110。但是,系统介绍多酸的电催化水裂解制氢的综述尚未见报道。本文将主要围绕多酸在电催化水裂解析氢的应用进行系统介绍,帮助读者了解多酸在电催化水裂解方面的研究进展。本综述先对电催化HER这部分的基本原理和存在问题展开叙述;随后,系统介绍多酸催化剂及作为催化剂前驱体在HER方面的应用研究进展;最后对多酸催化剂及作为催化剂前驱体的结构设计调控和提高电催化水裂解制氢性能方面进行展望。
2 电催化HER基本机理
在电解水的化学反应中,析氢反应发生在阴极,它是氢离子得到2个电子生成氢气的反应。其反应式如下:

早在1905年,Tafel便提出了电解水或酸性电解质溶液中析氢的重要步骤是氢原子的生成与重组111。Conway等112指出,酸性溶液中,在催化剂的存在下,电催化产氢(HER)分为以下三步反应:
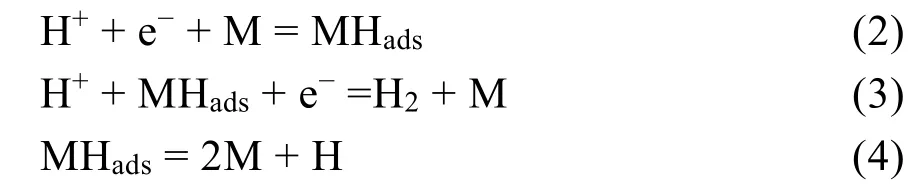
注:M代表催化剂的催化位点,Hads代表吸附的氢原子。
这三步反应分别为:伏尔默过程(the Volmer step)、海洛夫斯基过程(the Heyrovsky step)、塔菲尔过程(the Tafel step)。第一种是伏尔默反应,即初始电子转移过程,质子得到电子在催化剂表面生成吸附态的氢原子(Hads)。第二种是海洛夫斯基过程,该过程是由伏尔默反应生成的MHads进一步和电解质中的质子结合,同时获得一个电子之后,完成析氢反应的电化学脱附过程;第三种是塔菲尔过程,该步属于复合脱附过程,是由两个第一步生成的MHads复合生成氢气完成析氢反应的过程。
HER过程中,通常氢气产生的起始电位与φ(H+/H2)理论电位的差值,这部分电势称之为过电势(η),其大小与电极的材料有关。经过计算,全解水所需电压的理论值为1.23 V113,但由于过电势的存在,实际所需电压会大于此理论值。实验上,一般通过测试线性扫描伏安(LSV)曲线来评估催化剂的电催化HER性能。LSV反映了响应电流密度随不同电势下的变化。而且可以通过LSV进一步转化(以电流密度的对数为横坐标,过电势绝对值为纵坐标作图)可以获得塔菲尔曲线。一般在较高电流密度区间计算塔菲尔斜率。塔菲尔斜率是一个反映电极材料的催化动力学过程的重要数据。电催化析氢反应主要包括电子转移和氢气的脱附这两个过程。伏尔默过程反映了电子转移过程,该过程的塔菲尔斜率通常接近120 mV·dec-1,如果一个催化剂的塔菲尔接近该值,说明该催化剂的HER的决速步是电子转移过程,被称为“迟缓放电机理”;海洛夫斯基和塔菲尔过程反映了氢气脱附过程,这两个过程的塔菲尔斜率分别约等于40和30 mV·dec-1。如果一个催化剂的塔菲尔斜率约为40 mV·dec-1,说明该催化剂的决速步是由伏尔默反应生成的MHads进一步和电解质中的质子结合实现氢气电化学脱附的过程,被称为“电化学脱附机理”,经历的是伏尔默-海洛夫斯基(Volmer-Heyrovsky)过程;如果塔菲尔斜率约为30 mV·dec-1,说明复合脱附是反应中的决速步,称之为“复合脱附机理”,经历的是伏尔默-塔菲尔(Volmer-Tafel)过程。而实际的电催化过程的塔菲尔斜率往往介于这三个塔菲尔斜率之间。一般可以根据塔菲尔斜率的数值和交换电流密度来推测催化剂的电化学反应动力学过程114,115。理想的催化剂是具有较低的塔菲尔斜率和较高的交换电流密度。
如上所述,电催化析氢反应过程中,氢原子在催化剂表面进行着先吸附后脱附的反应。如果催化剂对氢原子吸附太强,能够很好地完成第一步电子转移的伏尔默反应和氢气的形成,但不利于氢气的脱附。若催化剂对氢原子的吸附太弱,则不利于形成氢气。只有在催化剂对氢原子的吸附和脱附能力达到良好的平衡,方可使催化剂表现出最佳的析氢活性。我们通常用氢吸附自由能(ΔGH)来表征催化剂的催化活性116。ΔGH越接近于零,析氢反应更容易进行。实验和理论结果计算表明:常见金属基催化剂的氢吸附自由能ΔGH和电催化HER活性呈“火山型”分布12,116。而铂系金属有着适中的氢吸附自由能,其ΔGH则位于金字塔的顶端,说明铂系催化剂具有极好的电催化析氢活性。因此,我们可以先通过理论计算方法计算不同材料的氢吸附自由能ΔGH,筛选ΔGH接近于零的催化剂,并进一步通过实验的方法将其制备出来用于电催化HER研究。
值得一提的是,Schmickler等117认为使用volcano plots (也称Sabatier principle)作为HER催化剂设计和优化的指导原则需要慎重。他们根据自己的理论研究了控制反应速率的因素,发现volcano plots只是决定速率的几个因素之一。尤其是对于氧化物覆盖的金属(如W、Mo、Nb、Ti、Ta),因为这些金属在电催化析氢反应中,容易在金属表面形成很薄的氧化层,而在以前做理论计算时并没有被考虑进去。而考虑金属被氧化物薄膜覆盖进行理论计算是相当困难的,因为这些薄膜结构很难确定,其往往是以多晶形式存在,甚至是无定型的;而且对氢的吸附形式于单质金属不同,其往往是以氧化物吸附质子形式存在;再者,金属和氧化物薄膜之间的电荷传输很难定量确定。这目前也无法通过实验的手段证实。除了镍和钴之外,对于氢吸附能力较强的金属,HER反应可能通过更合适的中间状态进行,其不会像Sabatier principle描述的那样导致反应速率降低。Schmickler等117认为,对于HER而言,不能简单考虑氢吸附自由能,其他指标,如功函数、晶格常数、熔化潜热等也应该被考虑进去。
对于析氢反应的机理,除了上述讨论的Volmer-Heyrovsky-Tafel (V-H-T)机理外,还有学者提出分子催化机理。下面我们将介绍一些学者在多酸分子的电催化析氢反应机理研究工作。Sadakane和Steckhan118认为多酸分子的电催化反应主要可以分为以下两种:(1)多酸溶解在电解质溶液中发生的均相反应;(2)多酸负载在电极表面发生的非均相反应。这两种方式并非完全独立,电催化反应过程中这两种反应有可能同时存在,因为溶液中的多酸在电催化反应过程中可能会沉积电极表面,同时,电极表面结合的多酸也会重新溶解在电解质溶液中参与电催化反应。他们认为多酸分子的电催化产氢机理与传统的吸附-脱附机理不同,在间接的电催化反应中,多酸可以作为一种电子传输的介质,首先溶液中的多酸分子在电极表面结合电子被还原成杂多蓝,然后和水反应生成氢气并回到原来的状态。
Savinov等119的研究成果可以佐证这一设想。他们利用锌汞齐还原硅钨酸H4[SiW12O40] (简称[SiW12]4-)获得杂多蓝,研究其在硫酸溶液中的析氢行为,并提出了多酸的HER可能机理如下:
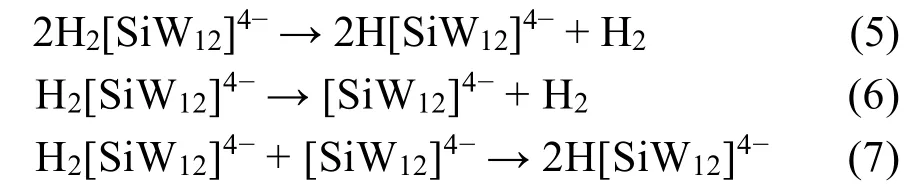
他们研究发现这种2电子还原的多酸H2[SiW12]4-是主要的HER活性物质,在酸性介质中,可以自身发生氧化还原反应生成氢气。然而,氢气的来源是由于还原剂锌汞齐中的锌单质将质子还原生成的还是2电子还原态多酸通过自身发生氧化还原反应生成的仍有待进一步探索。
此外,Akid和Darwent120还研究了[SiW12]4-的光催化HER性能,他们研究发现,只有在Pt存在下,多酸才能高效地将质子还原成氢气。如果没有Pt存在的话,仅有痕量的氢气产生。这说明Pt在多酸的光催化析氢反应中可以有效协同杂多蓝将质子还原成氢气。他们提出了以下反应机理:
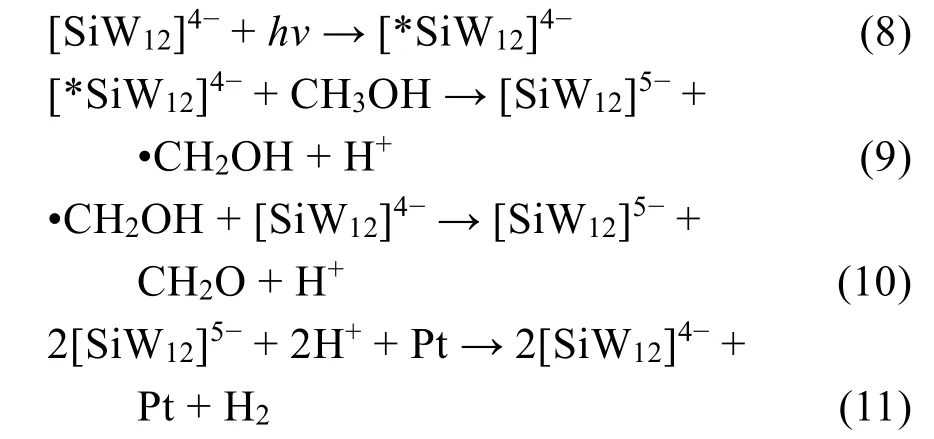
后来,Akid和Darwent121系统研究了硅钨酸[SiW12]4-沉积在不同电极材料表面的电催化析氢性能,他们研究发现,通过多次电化学沉积[SiW12]4-后,包括玻碳电极在内的多种电极表面都可以表现出与Pt接近的HER性能。不过,Sadakane等118也指出,通过电化学沉积的多酸对电极的电催化产氢性能较大促进作用也有可能与少量铂的沉积有关。
3 电催化HER存在的问题
铂碳是目前最高效、最稳定的商业化HER催化剂,然而铂在地壳中的储量较低,价格比较昂贵,限制了其大规模的工业化应用122,123。而石墨电极虽然成本低廉,但是其过电势非常大,效率很低,能耗较大,即从长远看来并不能达到节约成本的目的。所以,导电碳材料往往被用作导电添加剂或者电极支撑材料。目前我们需要寻找廉价又高效的HER催化剂。为了尽量降低HER过程中的成本,我们必须开发出高效的非贵金属析氢催化剂,在降低催化剂成本的同时,尽可能降低过电势导致的能耗。因此,绝大多数的文献都是基于以下六个因素考虑:(1)选用非贵金属催化剂来替代铂基催化剂或者降低铂的使用量降低催化剂成本;(2)提高催化剂的单位催化位点的活性来降低起始过电位,使其尽可能接近于零(相对于可逆氢电极RHE);(3)增加催化基活性位点的密度,使其在满足所需电流密度工作下降低过电势;(4)提高催化剂的导电性,降低欧姆电阻,提高电子传输能力;(5)改善催化剂和电极的界面,避免催化剂在反应过程中脱落,导致性能下降;(6)提高催化剂的稳定性21。
4 多酸在电催化HER的应用
多酸具有良好的氧化还原能力,可以可逆地接受和释放一个或多个电子124。而电化学反应正是通过电子转移发生,所以这为其作为HER催化剂提供了可能125,126。并且,多酸可以通过共价键与有机体系结合为多酸基无机-有机杂化化合物,还可以取代/掺杂其他元素,形成的杂多酸纳米团簇,这样可以对多酸的电化学等性质进行一定的调节127,128。并且,通过一些手段使多酸与其他物质结合为复合材料也是调节多酸本身性能的手段,这样可以改善多酸的导电性、溶解性和稳定性的问题129。
1985年,Keita和Nadjo121首次系统报道了多酸的电催化产氢性能。他们研究发现:硅钨酸(H4SiW12O40,简称:SiW12)在0.5 mol·L-1H2SO4电解质溶液中能被还原并修饰在玻碳(glass carbon,GC)表面,随着循环伏安扫描次数的增加,电极的电催化析氢活性进一步提高。说明这种还原态的SiW12是具有良好的电催化析氢的活性。而且,这种还原态的SiW12多酸可以在Pt的催化下高效地产生氢气120。随后,Nadjo等130利用共聚焦显微镜技术和电化学方法对多酸电催化剂的局部酸碱度和储存电子能力开展研究,并提出了“微环境效应(microenvironment effects)”对电催化析氢的影响,他们系统研究了[H7P8W48O184]33-(简称:P8W48) , [Co6(H2O)30(Co9Cl2(OH)3(H2O)9(β-SiW8O31)3)]5-( 简 称 : Co15)和 [(Co3(B-β-SiW9O33(OH))(B-β-SiW8O29OH)2)2]22-(简称:Co6)这三类多酸的电催化析氢性能。通过这三类多酸对玻碳电极的修饰和循环伏安电化学活化,可以有效提高电催化析氢性能,其中Co15表现出最佳的电催化析氢性能,其交换电流密度和塔菲尔斜率都非常接近商业Pt/C催化剂。因此,我们认为通过设计多酸的结构来调节其微环境,进而改善其捕获质子和电子的能力,提高多酸的电催化析氢性能是值得研究的一个方向。
多酸也可以和贵金属结合,协同作用于电催化析氢反应。2010年,Biboum等131利用K9[H4VPW17O62]与K2PdCl4的氧化还原反应和自组装行为,获得了具有“大型球形黑莓结构”的Pd0@POM,半径约为30-50纳米。反应中,V由+4价变为+5价,Pd由+2价变为0价的纳米粒子。该材料具有52 mV·dec-1的塔菲尔斜率,达到20 mA·cm-2电流密度时,需要300 mV的过电势。2011年,Barsukova-Stuckart等132报道了一例由22个钯稳定的二核铜,Na20[Cu2Pd22P12O60(OH)8]·58H2O,该团簇表现出电催化析氢的性质,其塔菲尔斜率为65 mV·dec-1。值得一提的是,2013年,Cronin等利用还原态SiW12多酸可以在Pt催化下快速地释放氢气,设计了电催化产氢和产氧的空间和时间的分离,可以有效避免电解水过程中氢气和氧气的混合,造成危险。他们通过电解获得氧气和还原态的SiW12多酸。然后将获得的还原态的SiW12转移至另一个容器中,在需要氢气的时候和Pt接触即可产生氢气125,126。2015年,美国北卡罗来纳大学的林文斌等133研究发现SiW11和Pt共沉积的方法获得了SiW11/Pt修饰的玻璃碳电极具有很好的HER催化性能(图1所示)。此电极中Pt的掺杂量极少,只有50-100 ng·cm-2。在10 mA·cm-2的电流密度下,其过电势为28 mV,塔菲尔斜率为32 mV·dec-1。
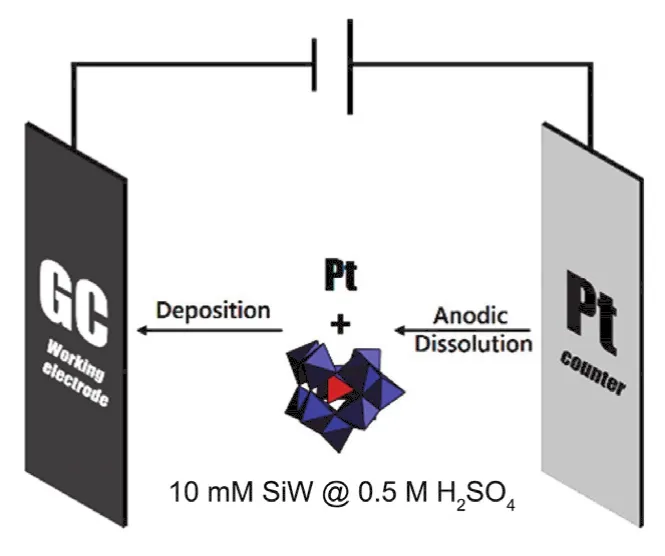
图1 Pt/POM催化剂电化学沉积在玻碳电极表面Fig.1 Electrodeposition of Pt/POM HER Catalysts on GC Surfaces.
多酸还可以和铜基材料结合用于电催化析氢反应。2018年,哈尔滨理工大学的马慧媛和庞海军课题组134将多酸装入Cu结点的metal organic frameworks (MOF)构成的金属有机纳米管骨架(MONT)中,获得了含多酸的HUST-200与HUST-201。这些特定的孔道结构有利于质子的传输。如图2,其中50% HUST-200表现出较好的HER性能,实现10 mA·cm-2的电流密度需要131 mV的过电势,塔菲尔斜率达到51 mV·dec-1。他们最近又合成了六钨酸阴离子和铜配位聚合物阳离子复合物,[Cu2(bimb)2(ox)](W6O19)·4H2O。其中50%质量分数的该材料和导电碳XC-72R复合物表现出最佳的电催化析氢性能,在10 mA·cm-2的电流密度下,其过电势为146 mV,塔菲尔斜率为69 mV·dec-1135。最近,清华大学魏永革教授课题组136将系列Anderson型杂多酸与铜电沉积在TiO2纳米管阵列上,制备了NiMo6O24@Cu/TNA、CrMo6O24@Cu/TNA和AlMo6O24@Cu/TNA三种多酸修饰的钛电极用于电催化析氢。我们研究发现,NiMo6O24@Cu/TNA表现出最佳的电催化析氢性能,塔菲尔斜率达到89 mV·dec-1,且相对于Cu/TNA材料的HER性能有较明显提升。
上述的研究说明了还原态的多酸是一类良好的电催化析氢催化剂,但是绝大多数的多酸容易结晶,得到的是大尺寸的晶态化合物。石墨烯是一种优秀的“二维矩阵(2D-Matrix)”,它可以固定各种分子催化剂,并且本身也具有优秀的电催化性能137。为了提高多酸活性位点的密度和电子传输能力,2016年,中国科学院过程工程研究所的张光晋研究员、德国不来梅雅各布大学的Kortz教授和东北师范大学的颜立楷教授等课题组合作报道了一种由48个钨和8个磷构成的还原态的大环杂多酸[H7P8W48O184]33-(P8W48)和还原石墨烯复合的电催化剂。他们是先利用电化学还原的方法获得在石墨烯表面均匀分散的还原态的P8W48多酸,然后部分还原态的P8W8多酸可以将氧化石墨烯还原为导电性良好的还原石墨烯,这样可以有效提供催化剂的电子传输能力(图3)129。该复合材料对比于每一单独组分的性能有很大增强。该研究通过吸附能与电荷转移的计算表明,P8W48与rGO (Reduced Graphene Oxide) 的具有较强的相互作用。该催化剂在酸性条件下具有比商业Pt/C优越的电催化HER性能。经过电化学测试,在10 mA·cm-2的电流密度下,其过电势仅30 mV,优于20%Pt/C电极。2018年,Fernandes等138进一步系统研究几种多酸-石墨烯复合材料(P2W18@rGF_ox、P5W30@rGF_ox、P8W48@rGF_ox)的电催化HER性能。其中,rGF_ox为还原氧化石墨烯片。它们的电催化HER性能也十分接近于铂,在电流密度为10 mA·cm-2时过电势仅35、33和44 mV。同年,Streb和Mirzaei课题组139首次报道了稀土金属掺杂对硅钨酸电催化析氢性能的影响。他们先合成了3例由3,4-吡啶二甲酸稳定的镧系金属取代的硅钨酸([LnSiW11O39]5-),分别为{LaW11}、{DyW11}、{PrW11},结构如图4所示。之后,将它们通过电化学还原沉积在rGO上,合成了{LaW11}@rGO、{DyW11}@rGO、{PrW11}@rGO。当实现电流密度为3 mA·cm-2时,需要过电势分别为0.14、0.17和0.41 V,塔菲尔斜率分别为71、86和185 mV·dec-1。而对没有稀土掺杂的{W11}@rGO,所需过电势为0.54 V,塔菲尔斜率为87 mV·dec-1。可以看出{LaW11}@rGO、{DyW11}@rGO中镧系金属元素对多酸的掺杂可以有效提高其电催化析氢的性能。

图2 晶态MOF材料的HER性能Fig.2 The HER performance of crystalline MOF materials.
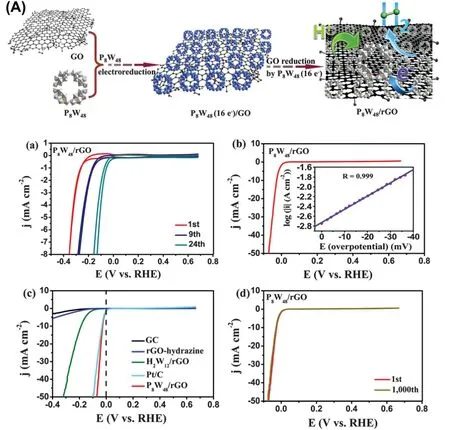
图3 P8W18/rGO催化剂的制备和HER性能Fig.3 The preparation of P8W18/rGO catalysts and their HER performance.
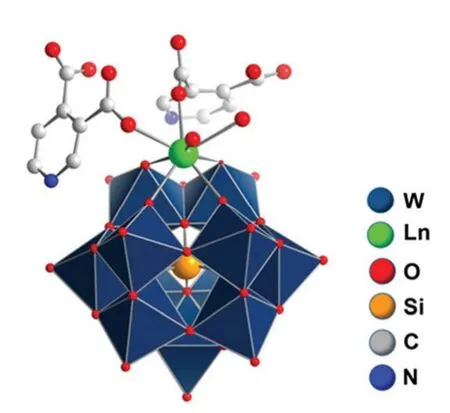
图4 镧系元素功能化的杂多酸[LnSiW11O39]5-Fig.4 Lanthanide-functionalized lacunary polyoxotungstates [LnSiW11O39]5-.
通过引入碳纳米管,同样可以提高多酸电催化析氢催化剂的电子传输能力,而且引入有机阳离子型聚合物可以有利于多酸阴离子纳米团簇的分散性。Ensafi等140,141将多酸负载在聚二烯丙基二甲基铵修饰的多壁碳纳米管(PDDA-CNT)上,再利用氯铂酸钾进离子交换,获得铂修饰的[PW11Pt-NiO39]@PDDA-CNT/GCE,该纳米复合材料表现出良好的电催化产氢性能。随后,该团队又报道了[PW11CoO39]5-@Ru-rGO、[PW11NiO39]5-@Ru-rGO、[PW11CuO39]5-@Ru-rGO142。这些材料的一大优点是在酸性及碱性条件下都可以稳定存在,其中含Co元素的材料性能相对与Ni、Cu更加优秀一些。
近年来,因为MOFs具有限域作用和比表面积大而且中心结点金属和配体可调等优点,也被广泛应用于电催化HER研究。其中多酸和MOF结合便是一种常见的思路。2015年,东北师范大学的苏忠民教授、南京师范大学的兰亚乾教授和德克萨斯A&M大学的周宏才教授课题组合作143报道了两例新颖含多酸纳米团簇的MOF材料,(TBA)3[ε-PMoV88MoVI4O36(OH)4Zn4](BTB)4/3和 (TBA)3[ε-PMoV8MoVI4O37(OH)3Zn4](BPT),分别命名为NENU-500和NENU-501。这两个多酸MOF (简称POMOF)在很广的pH (1-12)范围内都能稳定存在。他们研究发现:MOF中包含多酸可以提供HER的活性位点;而且多酸作为MOF材料的结点HER性能可以有效暴露出更多活性位点;而且,使用导电碳XC-72R复合可以有效提高材料的电子传输能力,进而提高电催化HER性能。因此,使用NENU-500掺杂的导电碳XC-72R作为电催化剂的HER性能比用单纯的NENU-500和单独的导电碳XC-72R要好的多144。这两例多酸MOF材料在0.5M H2SO4电解质溶液中,循环工作2000次后仍能保持良好的HER性能。其中最佳的NENU-500催化剂实现10 mA·cm-2电流密度所需过电势为237 mV,塔菲尔斜率为96 mV·dec-1。最近,东北师范大学的李阳光教授等145合成了一种由Zn、P、Mo(VI)和Mo(V)构成的杂多酸节点与1,4-二(1-咪唑基)苯配体连接形成的POMOF,他们利用该POMOF含有还原态的五价Mo(V)能将H2PtCl6还原为零价的Pt,进而修饰在POMOF表面,形成Pt@POMOF-1,通过掺入导电碳KB,Pt@POMOF-1/KB表现出优异的电催化HER性能,过电势非常接近于20% Pt/C电极。这种方法可以很好地降低Pt的使用量,实际上只掺杂了0.43% (w)的Pt,可以极大节约成本。
便于查阅,我们将部分代表性的多酸催化剂HER性能整理至如下表1。
5 多酸作为前驱体在电催化HER的应用
得益于还原态的Mo和W多酸及其衍生物具有良好的电催化析氢活性,被广泛应用于电催化HER研究。但是多酸自身导电性较差,作为HER电催化剂,需要引入额外的导电碳基底以提高其电子传输能力。除了还原态多酸电催化剂,近几年来,科研工作者研究发现低价钼或钨的硫化物146-155、碳化物156-162、氮化物163,164、磷化物165-172和氧化物3,41,44表现出很好的电催化析氢活性。然而,由于这些材料一般都是通过高温焙烧获得的,在高温焙烧过程中,很难实现均一的碳化、氮化和磷化等,而且很难避免颗粒的团聚。
自从2012年,瑞士洛桑联邦理工学院的胡喜乐教授等158首次系统研究了碳化钼和硼化钼的电化学析氢性能,他们发现无论在酸性还是碱性介质中商业购买的β-Mo2C微粒均表现出超高的电催化产氢活性和稳定性29。迄今为止,商业上生产碳化钼的方法主要有CH4/H2混合气还原法173、有机金属钼化合物的热解法174、微波辅助热分解法175和化学气相沉积法(CVD)176。因为多酸结构明确,具有纳米级的尺寸,而且含有丰富的钼和钨原子,被认为是良好的钼和钨基催化剂的前驱体。研究表明钼酸铵((NH4)6Mo7O24·4H2O)可以作为前驱体,为碳化钼纳米材料的制备提供丰富的钼源。Chen等2发现将钼酸铵((NH4)6Mo7O24.4H2O)和伏尔甘XC-72R炭黑在水中混合均匀,然后130 °C烘干,随后在氩气中进行800 °C高温热解2 h,获得碳化钼纳米材料Mo2C/XC-72,如果加入碳纳米管即可获得碳纳米管负载的碳化钼纳米材料Mo2C/CNT (图5)。研究表明,利用该方法获得的碳化钼纳米材料的电催化析氢性能比购买的碳化钼材料好,而且用碳纳米管负载的碳化钼纳米材料具有更好的电催化产氢活性和稳定性。2014年,上海复旦大学的唐颐教授和刘宝红教授等利用钼酸铵提供钼源、苯胺提供碳源,调节pH为4-5,通过盐酸酸化处理得到前驱体Mo8O26(C6H8N)4·2H2O,将前驱体在氩气的条件下进行高温725 °C热解5 h,即可获得碳化钼纳米线np-Mo2C NWs,该碳化钼纳米线具有丰富的孔道结构,能够暴露出较多的活性位点,而且表现出非常好的电催化产氢性能,析氢初始电位为70 mV,在过电位为200 mV时,能获得60 mA·cm-2的电流密度,其性能远远超过块状的碳化钼材料。
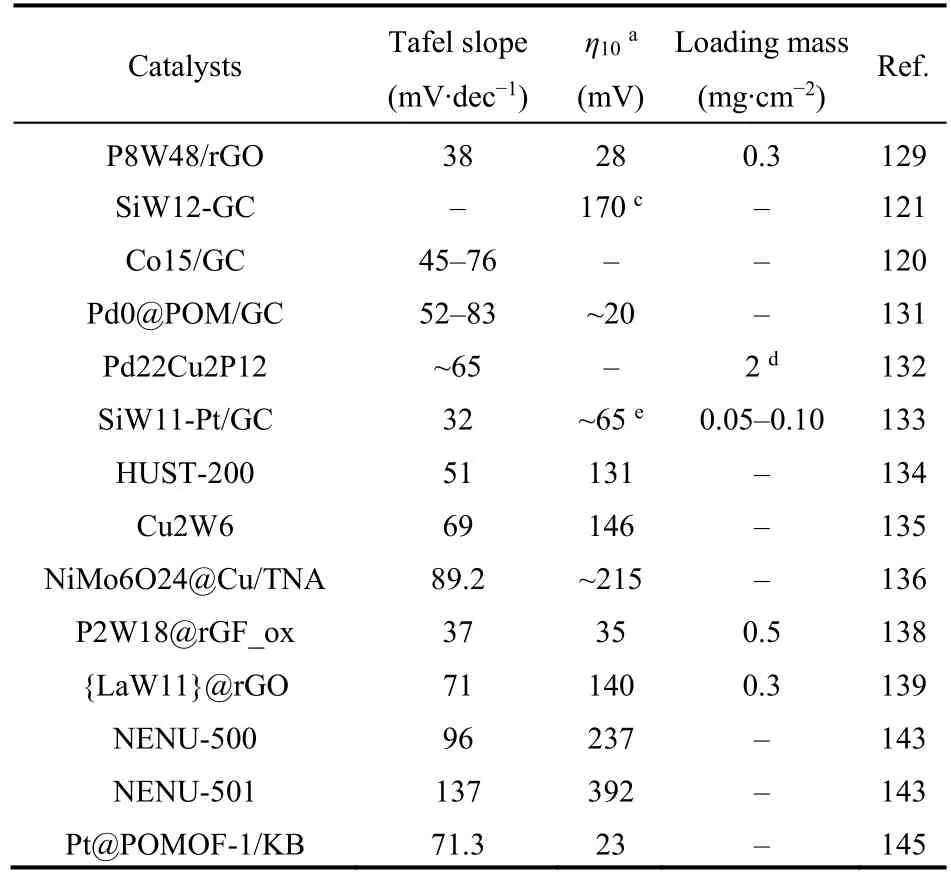
表1 多酸基电催化剂的HER性能比较Table 1 Comparison of HER performance for POM-based electrocatalysts.

图5 Mo2C/XC和Mo2C/CNT催化剂的形貌和HER性能测试Fig.5 The morphology of Mo2C/XC and Mo2C/CNT catalysts and their HER performances.
最近,兰亚乾等4利用Keggin型多酸PMo12、聚吡咯、还原石墨烯三元体系作为前驱体,经过900 °C高温N2焙烧,获得二维耦合的碳化钼材料杂化材料,由N,P掺杂石墨烯和N,P掺杂的碳壳封装而成,简称:Mo2C@NPC/NPRGO。如图6所示,碳化钼颗粒很好地分散在还原石墨烯上,而且N和P元素均匀的掺杂在还原石墨烯上面,提供更多的活性位点。该材料表现出优异的电催化析氢性能,在0.5 mol·L-1的硫酸水溶液中,实现10 mA·cm-2的电流密度,仅需34 mV的过电势,塔菲尔Tafel斜率为33.6 mV·dec-1,而且循环了1000次,其电催化析氢活性几乎没有变化。此外,碳纳米管也可以增强电催化剂的导电性,而且与石墨烯复合能起到协同作用,在增强导电性的同时提高催化剂颗粒的分散性,有效避免颗粒的团聚,暴露出更多的活性位点。2014年,Lee课题组分别将Mo2C、Mo2N和MoS2负载在碳纳米管和石墨烯(CNT-Graphene)的基底上,获得以CNT-石墨烯基底的高分散性的纳米晶材料177。通过电催化性能研究发现,Mo2C/CNTGraphene材料表现出最好的催化析氢活性,起始电位为62 mV,塔菲尔斜率为58 mV·dec-1,而且具有很好的循环稳定性。
中山大学的匡代彬教授和苏成勇教授等53以钼酸铵和钨酸钠为原料,通过水热控制的Mo-W氧化物在碳布上生长,获得均一的纳米线,然后在高温下对该纳米线进行原位磷化,成功制备磷和钨掺杂的纳米片状的Mo-W-P双金属材料。该材料具有非常好的电催化析氢性能,在0.5 mol·L-1的硫酸水溶液中,实现100 mA·cm-2的电流密度,仅需138 mV的过电势,塔菲尔Tafel斜率为52 mV·dec-1,而且具有较好的循环稳定性能。但是从文章提供的透射电镜(transmission electron microscopy,TEM)图和扫描电镜(scanning electron microscopy,SEM)图来看,虽然形貌比较均一,但其获得Mo-W-P材料仍有团聚现象。如果能改善其团聚现象,有望进一步提高其性能;同时该材料无法实现严格的定量掺磷和调控钨的比例,如果能够从前驱体入手实现定量掺磷和调控钨掺杂比例,有望优化该系列催化剂的性能,获得性能更好的催化剂。
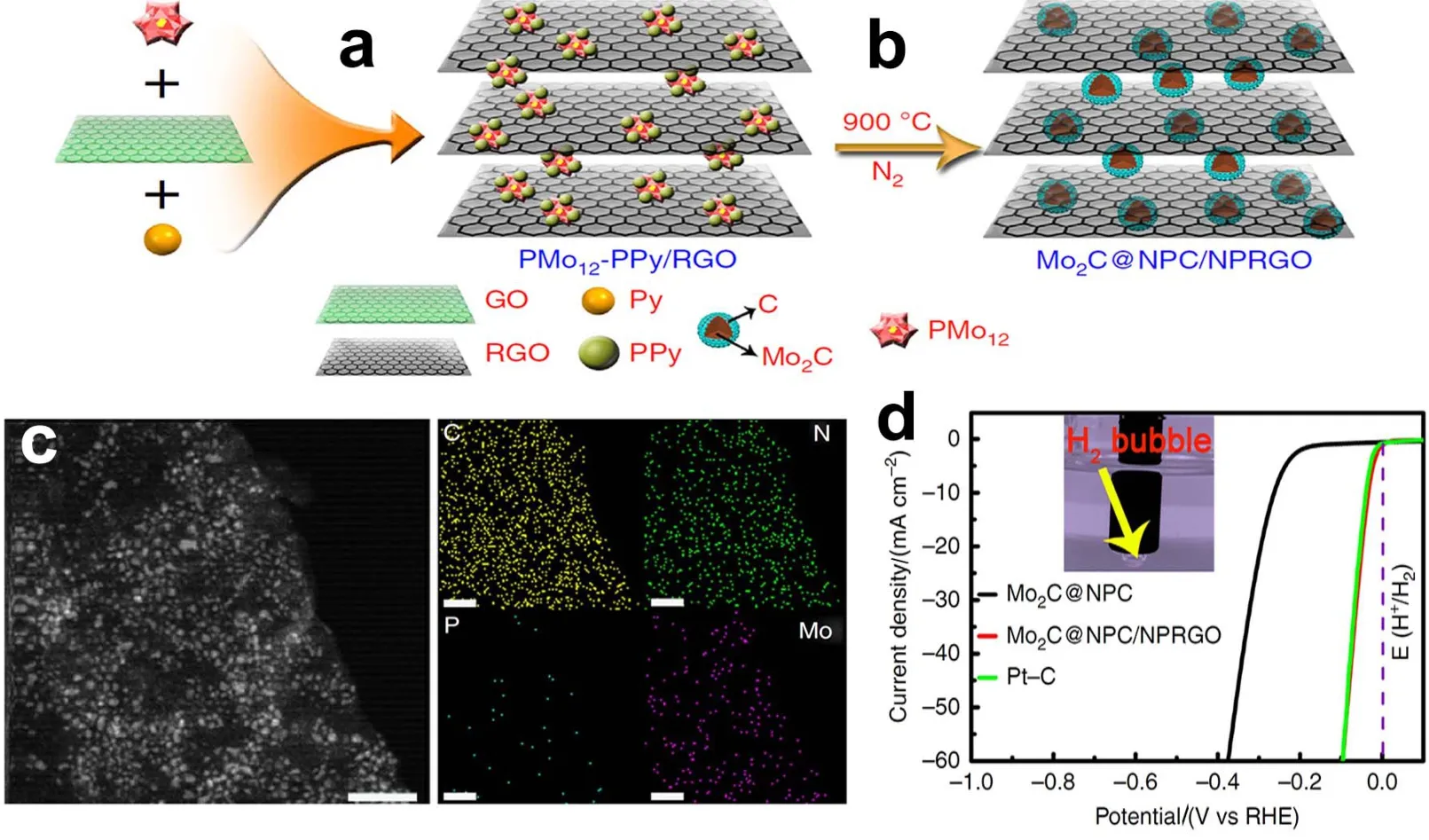
图6 Mo2C@NPC/NPRGO催化剂的制备和形貌表征以及HER性能Fig.6 The preparation, morphology characterization and HER performance of Mo2C@NPC/NPRGO catalysts.
最近,相关研究表明多酸-MOF复合材料可以作为前驱体,制备多孔的碳化钼纳米材料。同时,MOF材料的桥连有机配体可以作为碳源,在高温焙烧过程中原位生成导电碳,也可以有效避免纳米颗粒的团聚。中国科学技术大学的俞书宏教授和南京师范大学的兰亚乾教授178等利用杂多酸H3PMo12O40·nH2O、Cu(OAc)2·H2O、H3BTC和氧化石墨烯(GO)合成POMOFs/GO前驱体,通过800 °C条件下热解3 h获得MoO2@PC-rGO材料。如图7所示,该材料的前驱体由综合了POM、MOF和GO的优点。用3 mol·L-1HCl洗涤除去铜,最终得到了多孔的MoO2@PC-rGO。电流密度为10 mA·cm-2时,需要过电势为64 mV,塔菲尔斜率为41 mV·dec-1。后来,兰亚乾和徐强课题组合作报道了在导电碳布上负载含磷钼酸的铜基MOF,将其作为前驱体,经过高温磷化后获得Cu-Mo-P/CC等系列三相催化剂,研究结果表明,该催化剂在0.5 mol·L-1H2SO4、1 mol·L-1KOH、0.5 mol·L-1PBS不同电解质溶液中表现出良好的电催化析氢活性179。
2016年,南京师范大学的兰亚乾和戴志晖等1同样以多酸-MOF为前驱体合成的Fe3C/Mo2C@NPGC材料,该材料具有特殊的介孔结构,如图8所示。该材料在电流密度为10 mA·cm-2时过电势为98 mV,塔菲尔斜率为45 mV·dec-1,这电催化产氢性能比单独的Fe3C@C要优秀许多。2年后,该团队180又以另一种多酸-MOF(NENU-5)作为前驱体,经过600 °C烧结、以Fe3+除去Cu、以(NH4)2HPO2磷化得到MoP@PC/rGO。该材料在电流密度为10 mA·cm-2时过电势为235 mV,塔菲尔斜率为53.6 mV·dec-1。
楼雄文课题组同样利用MOFs辅助合成的方法设计合成含Keggin型多酸(PMo12)的MOF前驱体,经过高温热解获得多孔的碳化钼MoCx八面体纳米颗粒156。该材料表现出优异的HER催化活性,在0.5 mol·L-1H2SO4中,析氢起始电位为25 mV,电流密度为1和10 mA·cm-2时,所需的过电位为87和142 mV,塔菲尔斜率为53 mV·dec-1,交换电流密度为0.023 mA·cm-2。在1 mol·L-1的KOH中,析氢起始电位为80 mV,电流密度为1和10 mA·cm-2时,所需的过电位为92和151 mV,塔菲尔斜率为59 mV·dec-1,交换电流密度为0.029 mA·cm-2。
东北师范大学的李阳光、王新龙和谭华桥等人181设计了Zn作为结点含氮双咪唑类有机物作为配体,多酸作为的配位的MOF报道了氮掺杂的MoCx@C-1材料,其前驱体也是以POM为中心的POMOF。电流密度为10 mA·cm-2时过电势为79 mV,塔菲尔斜率为56 mV·dec-1。2017年,李阳光、康振辉和谭华桥106等合成了粒径为5-20 nm的CoMoP@C材料。该材料是以(Co16Mo16P24)多酸骨架与双氰胺(DCA)作为前驱体,800 °C烧结而成的。在反应中,DCA可以引导氮掺杂的碳的原位合成,将CoMoP@C包裹,以提高其稳定性。该材料在电流密度为10 mA·cm-2时过电势为41 mV,塔菲尔斜率为50 mV·dec-1。同年,该课题组182同样利用磷钨酸与DCA在400 °C条件下烧结得到PW2C@NC,在电流密度为10 mA·cm-2时,过电势为89 mV,塔菲尔斜率为53 mV·dec-1;2017年,他们183又以类似的方法合成了MoP/Mo2C@C。在电流密度为10 mA·cm-2时过电势为89 mV,塔菲尔斜率为45 mV·dec-1;该团队又于2018年合成了Co2P/WC@NC184、P-Mo2C@NC185和TMMo2C@C186(TM = Cr、Fe、Co、Ni)。它们同样具有优秀的电催化HER性能。
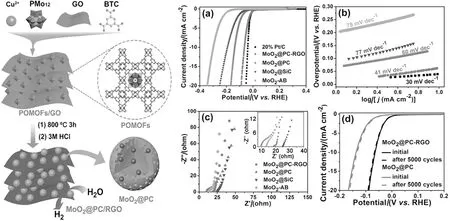
图7 MoO2@PC-rGO纳米复合材料的合成及其HER性能Fig.7 Preparation of the MoO2@PC-RGO nanocomposite and their HER performance.
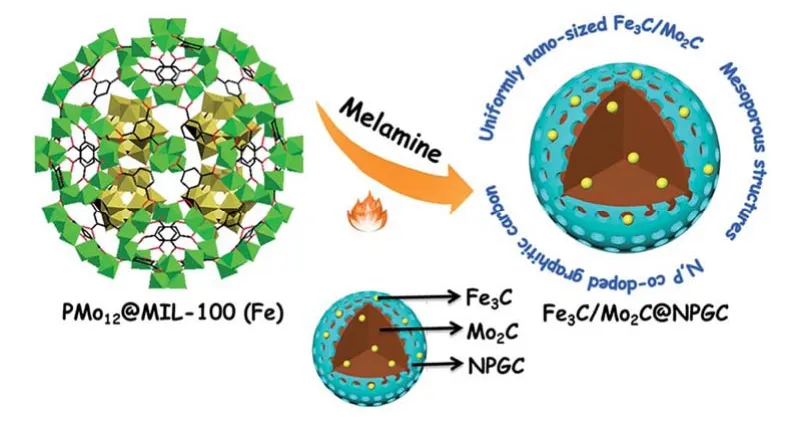
图8 Fe3C/Mo2C@NPGC催化剂的制备及其介孔结构Fig.8 Preparation and its mesoporous structure of the Fe3C/Mo2C@NPGC catalyst.
2018年,清华大学的魏永革教授课题组利用“DDC-脱水法”187,188设计合成了一个由2个苯基磷酸配体稳定的4核钼的杂多酸苯亚胺衍生物Mo4-CNP的分子作为前驱体,利用单晶解析技术获得Mo4-CNP的晶体结构,在晶体内部,Mo4-CNP分子之间通过π-π相互作用组装成纳米片32。通过高温惰性气体热解,形成多孔的氮掺杂的碳化钼和磷化钼纳米片杂化材料N@MoPCx(图9)。该材料表现出优异的电催化析氢活性和稳定性。起始过电势为32 mV,塔菲尔斜率为69.4 mV·dec-1。研究结果表明苯亚胺,苯基磷酸配体可以作为N、C、P源,在高温惰性气体条件下实现均匀的掺杂、碳化和磷化,从而生成氮掺杂的碳化钼和磷化钼杂化材料。4核钼簇周围被有机配体包围,有利于提高所生成碳化钼和磷化钼的分散性,暴露更多的活性位点;而且有机物高温热解过程中产生气体能有效提高材料的孔隙率,增强电催化传质过程,进而加速催化过程;另外,可以原位形成碳基底,提高催化剂的导电性。这种从多酸前驱体分子设计实现原位掺杂的方法,可以有效调控催化剂的电子结构,获得不同的催化剂复合材料32。
2019年,清华大学的魏永革教授课题组,圣地亚哥州立大学的顾竞教授课题组、北京化工大学的程道建课题组和浙江工业大学的张诚教授课题组合作报导了“多酸有机亚胺配体修饰”的策略(图10)制备可控氮掺杂的碳化钼,获得了高效的非贵金属析氢电催化剂。该策略的核心是通过原位构筑具有可调控Mo―N键的不同亚胺有机配体所取代的多酸前驱体,在惰性气氛高温热解获得不同氮掺杂程度的碳化钼纳米片,成功实现碳化钼的精准定量、定位可控氮掺杂。该工作从理论和实验上证实了该策略能有效地实现碳化钼的氮掺杂,适当的氮掺杂能够有效调整碳化钼的电子结构,降低Mo-H的结合能力,进而促进析氢过程,大幅度提高材料的电催化析氢活性105。
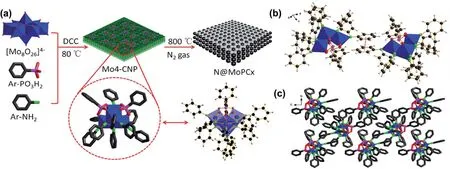
图9 多孔氮掺杂碳化钼与磷化钼的复合材料的制备和Mo4-CNP前驱体结构示意图Fig.9 The preparation of porous nitrogen-doped molybdenum carbide and phosphide hybrid and the structures of Mo4-CNP precursor.

图10 多酸有机亚胺化修饰策略构筑氮掺杂碳化钼析氢电催化剂Fig.10 The preparation of nitrogen-doped molybdenum carbides as HER electrocatalysts via the organoimido functionalization of POMs.
2018年,河南大学的牛景杨教授和王敬平教授课题组189合成了[Hx(MCp*)4W8O32]·yH2O (M =Rh,x= 8,y= 23;M = Ir,x= 4,y= 17.33),Cp代表五甲基环戊二烯基。然后将其负载在泡沫镍上作为前驱体,在400 °C,15% H2/N2混合气氛下烧结8 h,得到还原产物M0.4Ni0.6/WO3。电流密度为3 mA·cm-2时过电势分别为67 mV (Rh)、35 mV(Ir),塔菲尔斜率分别为56 mV·dec-1(Rh)和34 mV·dec-1(Ir)。2018年,中国科学院福建物质结构研究所的王瑞虎研究员课题组190以碳量子点CQD、磷钼酸、次磷酸钠为原料自组装为前驱体,高温热解合成了CQDs/MoP材料(CQD代表碳量子点),该工作首次将多酸与碳量子点结合,并发现了其优越的电催化析氢性能。在电流密度为10 mA·cm-2时过电势为210 mV,塔菲尔斜率为56 mV·dec-1(1 mol·L-1KOH)。
多酸除了可以作为碳化钼和磷化物电催化析氢材料,还可以通过硫化获得硫化钼纳米材料。2016年,南京师范大学的兰亚乾和李亚飞课题组将磷钼酸和石墨烯复合,通过加入硫脲在160-220 °C进行硫化后获得不同层间距的二硫化钼/还原石墨烯复合材料,其中在180 °C获得的样品MoS2/NRGO-180表现出最佳的电催化析氢性能。起始过电势为5 mV,达到10 mA·cm-2的电流密度需要56 mV的过电势,塔菲尔斜率为41.3 mV·dec-1,非常接近于商业Pt/C催化剂191。后来,Singh课题组192报道了利用硫化氢气体作为硫源,磷钼酸作为钼源,获得了二硫化钼和还原石墨烯的复合材料MoSx-rGO。其XRD、TEM和元素分析表明其主要含有MoS3纳米晶和少量MoS2纳米片。该材料在电流密度为10 mA·cm-2时过电势为176 mV,塔菲尔斜率为44 mV·dec-1。2019年,哈尔滨工业大学的邱云峰等193以磷钼酸的吡咯盐为前驱体,用单质硫在800 °C下进行硫化得到MoS2/C,用于电催化析氢反应。在电流密度为10 mA·cm-2时过电势为207 mV,塔菲尔斜率为73 mV·dec-1。
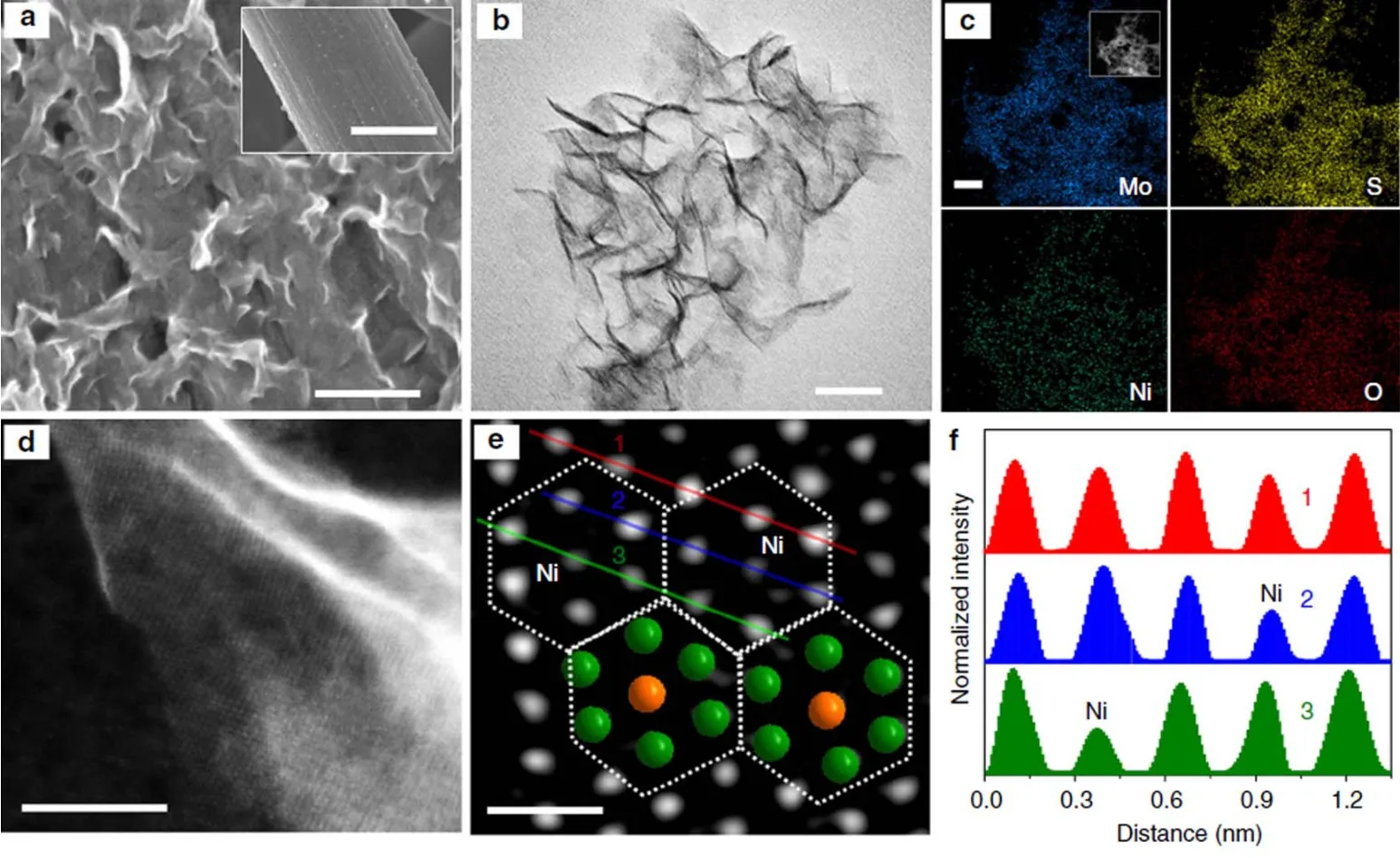
图11 NiO@1T-MoS2催化剂的结构表征Fig.11 Structure characterizations of NiO@1T-MoS2.
相对于稳定性较好的2H-MoS2而言,介稳态的1T相MoS2(1T-MoS2)具有较好的电子传输能力和更高的电催化析氢活性194-197。但是在反应过程中,1T-MoS2容易转化为2H-MoS2,导致电催化析氢性能下降。通过锂离子插层的方法可以将2HMoS2转化为1T-MoS2,但是这种方法产率较低,不利于大规模应用195,197。最近,国家同步辐射实验室的宋礼等人利用钼酸铵(NH4)6Mo7O24·4H2O (简称Mo7)作为前驱体与有机硫化合物反应,获得了高纯度和高稳定性的1T-MoS2194。考虑到Mo7是一种蝴蝶构型的结构,其结构与Anderson型多酸的β同分异构体类似82,清华大学魏永革教授课题组、美国圣地亚哥州立大学顾竞教授课题组和吉林大学张立军教授课题组合作提出利用Anderson-型杂多酸作为前驱体制备高纯度的1T-MoS2的研究思路。Anderson-型多酸是由一个杂原子XO6八面体(X = Fe、Co、Ni等)被六个共边的钼氧MO6八面体围绕连接而成198。Anderson-多酸独特的分子结构有望获得均一过渡金属掺杂的1T-MoS2,实现对1T-MoS2电子结构的有效调整,进而提供更多的活性位点。这个工作设计合成了一系列Anderson-型多酸,[XH6Mo6O24]n-(简写为XMo6;X = FeIII,CoIII,n= 3;X = NiII,n= 4),作为前驱体,以硫代乙酰胺作为硫源通过水热反应制备过渡金属和氧原子共掺杂的1T-MoS2(如图11所示),包括FeO@1T-MoS2、CoO@1T-MoS2和NiO@1T-MoS2材料21。其中,NiO@1T-MoS2表现出最佳的电催化析氢性能,在电流密度为10 mA·cm-2时过电势仅有46 mV,塔菲尔斜率为52 mV·dec-1。在这个工作发表之前,南京师范大学的兰亚乾教授课题组58和大连理工大学的孙立成教授课题组60先后利用Anderson-型多酸制备了高效的双金属硫化物/碳布复合电催化剂用于全解水反应。便于查阅,我们将上述代表性的多酸热处理后的电催化剂HER性能整理至如下表2。
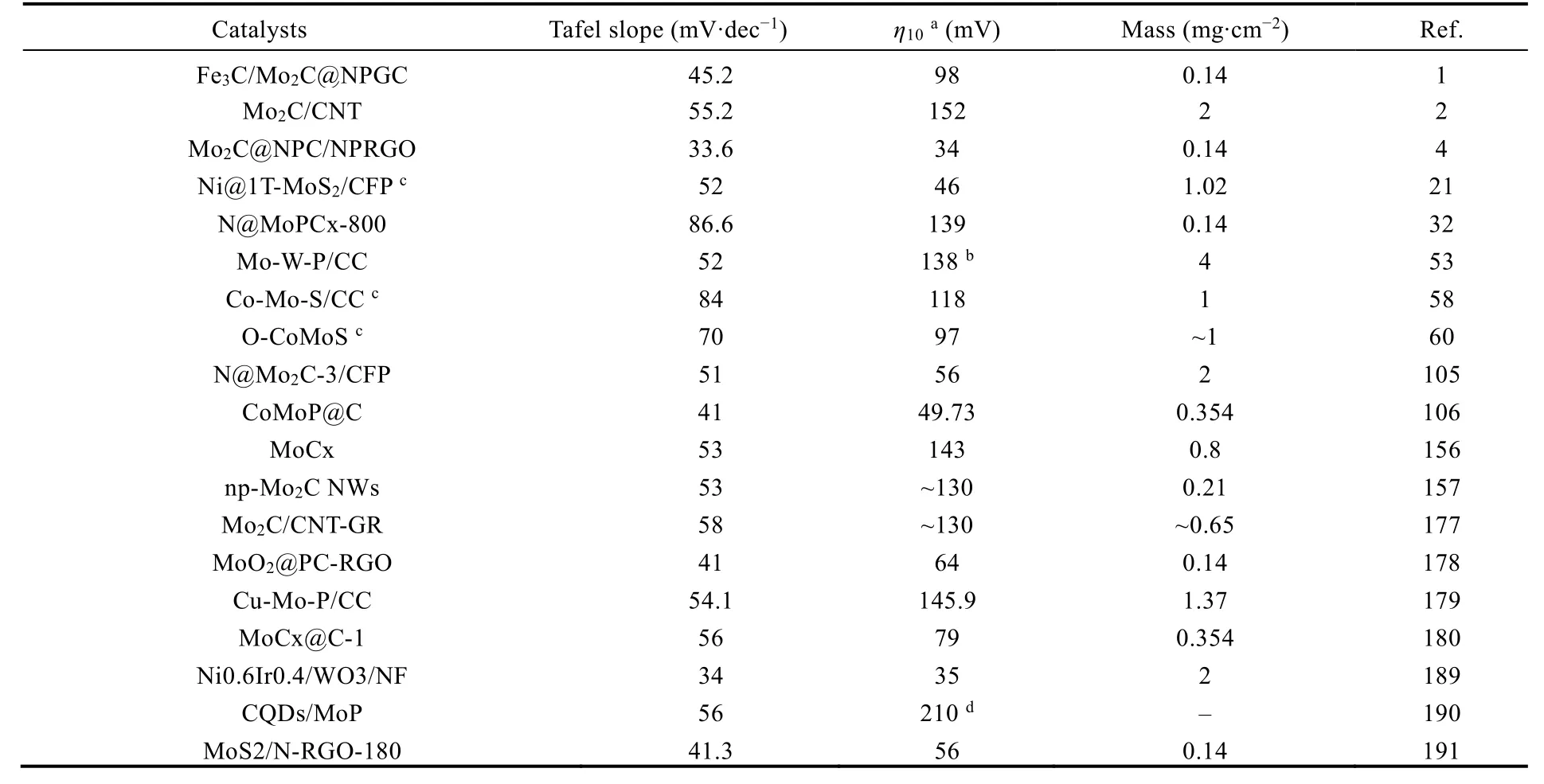
表2 多酸热处理后电催化剂的HER性能比较Table 2 Comparison of HER performance for pyrolyzed POMs electrocatalysts.
6 结论与展望
总的来说,还原态的多酸和高导电性的碳基材料(如石墨烯、碳纳米管、炭黑XC-72R或KB等)复合是获得高效电催化HER催化剂的有效途径。还原态的多酸含有低价金属是主要的催化HER活性位点,通过引入极少量的Pt可以有效提高其电催化HER性能121,132。但是毕竟Pt基贵金属价格昂贵,如果能完全使用非贵金属的HER催化剂的话,那将更具有吸引力。从目前报道的结果来看,通过过渡金属掺杂(Co、Ni和La等)还原态的多酸来调整多酸自身的氧化还原活性,进而提高多酸的HER性能的策略也是行之有效的。电催化析氢除了催化剂自身的活性,其电子传输能力也不容忽视。多酸自身导电性差,不利于电子传输,通过引入导电性良好的材料,可以有效提高催化剂的电子传输能力,进而提高其电催化HER性能。多酸直接用于电催化HER今后需要解决的问题主要有以下三个方面:一是如何实现多酸和导电材料的紧密结合,提高电子传输能力的同时提高催化剂的稳定性;二是提高多酸的分散性,避免多酸自身发生团聚,暴露出更多的活性位点;三是寻找HER活性更高、更稳定的多酸分子,提高其HER的本征活性。多酸电催化的pH效应也很重要,目前HER研究主要集中在酸性和碱性,因为中性条件下的离子传输性能较差,中性条件下的电催化HER研究尚少。一般来讲,在酸性条件下更有助于多酸催化剂的HER,因为多酸容易结合质子,进而在还原过电势的作用下进一步被还原生成氢气。而在碱性条件下,质子含量很少,传质受阻,需要先使水裂解产生氢离子或者氢原子再进一步产生氢气,需要消耗额外的能量。然而,碱性条件下有助于电催化产氧(OER),未来的发展方向需要开发出碱性条件下的高效HER电催化剂用于全解水反应,这对于多酸而言是非常有潜力的,因为多酸在碱性条件下容易发生水解结合质子。但是我们仍需要筛选出合适的耐碱性和氧化还原性能优异的多酸催化剂。
将多酸纳米团簇进行高温热解反应(碳化、磷化、氮化和硫化等)是提高多酸基电催化剂活性、导电性和稳定性行之有效的方法。但是也存在一些问题,多酸纳米团簇在高温热解过程中容易团聚,过渡金属取代的多酸纳米团簇热解后往往容易形成多相结构,导致活性位点不明确。将多酸纳米团簇装进金属有机框架(MOFs)材料中,获得多酸基MOFs (POMOFs)前驱体,然后将其热解是目前比较流行的方法。但是POMOFs一般尺寸较大,如果能够获得超细的POMOFs作为前驱体,将有望进一步提高电催化剂的析氢活性。还有一个思路是将多酸和含氮/磷/硫聚合物(聚吡咯、聚噻吩等)复合,以提高多酸的分散性,同时实现杂原子掺杂提高电催化剂的活性和导电性。此外,如何可控设计调控催化剂的活性位点,来提高催化活性的同时,提高催化位点的密度是提高电催化析氢性能的关键。将多酸纳米团簇表面进行有机修饰引入杂原子,可以实现电催化剂的可控杂原子掺杂,这个思路可以很好地调控催化剂的活性位点的单位活性和密度。
对于目前报道的电解水制氢催化剂有很多,主要集中在报导催化剂结构和形貌对其电催化HER活性的影响,但是对长时间的稳定性测试、以及催化过程中催化剂的结构以及活性位点的变化的报导为之尚少,而这些研究可以为我们提供重要的数据支撑和指导,对高效稳定的电解水制氢催化剂的开发至关重要。此外,我们还需建立一套针对电催化析氢反应HER研究的标准和规范,这将有助于我们开发出更适合商业化的电解水析氢催化剂。
猜你喜欢
油气田地面工程(2022年8期)2022-10-02
有色金属科学与工程(2022年1期)2022-03-12
学校教育研究(2021年20期)2021-12-14
防爆电机(2020年4期)2020-12-14
大连工业大学学报(2020年1期)2020-01-17
武汉工程大学学报(2019年5期)2019-11-02
分析化学(2018年12期)2018-01-22
汽车生活(2017年4期)2017-04-26
分析化学(2017年1期)2017-02-06
中小企业管理与科技·下旬刊(2016年12期)2017-01-17
