
SIRT2稳定敲除细胞系的建立及对组蛋白修饰的影响
2020-03-31 03:15刘长云
医学研究杂志 2020年2期
李 雪 裴 培 刘长云 王 珊
CRISPR/Cas9已被证明是一种简单有效的基因组编辑工具[1]。CRISPR-Cas系统(CRISPR-Cas system)可以进行多靶点基因编辑。CRISPR相关内切酶(CRISPR-associated,Cas)能够切断外源性的核酸,并能够由两种小分子RNA将其定向到其靶基因序列上[2]。据报道cas蛋白家族已经超过40多种,基于Cas蛋白的序列和结构,CRISPR/Cas系统主要分为Ⅰ、Ⅱ和Ⅲ3种类型[3,4]。Ⅱ型CRISPR/Cas系统仅需要单个Cas蛋白Cas9,其含有HNH核酸酶结构域和RUVC样核酸酶结构域[5]。该系统由两部分组成——tracrRNA和crRNA,两部分融合表达之后,形成一个具有引导功能的小向导RNA(small guide RNA, sgRNA)。sgRNA的5′端与目的DNA的20个核苷酸特异性结合,3′ 端可以结合核酸内切酶Cas9并将其激活,引导Cas9对目的DNA进行精确切割[6]。该技术因能更有效、更便捷地编辑基因而被广泛应用。
酵母沉默信息调节蛋白2(silent information regulators2, SIRT2),是一个高度保守的烟酰胺腺嘌呤二核苷酸(nieotinamide adenie diueleotide, NAD+)依赖的组蛋白去乙酰化酶(histonedeacetylase, HDAC)和ADP核糖基转移酶[7]。哺乳动物的sirtuins蛋白有7类,分别为SIRT1~SIRT7,它们具有高度保守的NAD+结合域和催化功能域,不同的N端和C端使具有不同的底物特异性和亚细胞定位[8~10]。该家族成员参与基因沉默、新陈代谢、组蛋去乙酰化、细胞周期调节和抗衰老等多项生物学过程[11]。SIRT2最初作为微管蛋白的去乙酰化酶被认识,进一步的研究表明,SIRT2可以去乙酰化很多重要的转录因子和调节蛋白,从而调控多种生物学过程[7,12]。SIRT2还可以对组蛋白进行修饰、使其去乙酰化,在细胞周期的有丝分裂过程中,SIRT2从细胞质进入细胞核并与染色质发生共定位。
本研究利用慢病毒结合CRISPR/Cas9技术将HEK293细胞中的SIRT2基因敲除,建立SIRT2稳定敲除的细胞系,为深入研究SIRT2对组蛋白的修饰功能提供基础。
材料与方法
1.实验材料:人胚肾细胞HEK293(中国医学科学院基础医学研究所国家实验细胞资源共享平台)。人源胚胎肾细胞变种T细胞、菌种DH5ɑ(本实验室保存)。0.25% Trypsin-EDTA、DMEM培养基、胎牛血清(美国Gibco公司)。PBS(美国Hylcone 公司)。细胞培养瓶、培养板及移液管等细胞培养耗材(美国Coring公司)。二盐酸嘌罗霉素(嘌呤霉素,puromycin dihydrochloride, 美国Amresco 公司)。lentiCRISPRv2质粒(美国Addgene公司);sgRNA寡核苷酸序列合成(中国碧云天生物技术研究所)。限制性内切酶BsmBI(日本TaKaRa公司)。DNA提取试剂盒(中国天根生化科技有限公司)。PSMF、SDS-PAGE凝胶试剂盒(北京索莱宝生物科技有限公司)。2×Loading Buffer(北京碧云天生物技术研究所)。BCA蛋白浓度定量试剂盒、蛋白Marker(美国Thermo Fisher Scientific公司);超敏ECL化学发光检测试剂盒、ECL化学发光检测试剂盒(美国Thermo Fisher Scientific公司)。GADPH抗体、SIRT2抗体(美国Proteintech公司)。辣根过氧化物酶(HRP)偶联山羊抗兔IgG(北京中杉金桥公司)、Acetyl-Histone H4(Lys5)抗体、Acetyl-Histone H4(Lys16)抗体、H3K79me2抗体(美国Cell Signaling公司)。
2.LentiCRISPR v2-sgRNA SIRT2敲除质粒的构建:人的SIRT2基因的Gene ID为22933,首先在NCBI中查找22933基因,找到该基因的CDS区,分析相应的基因组结构,明确CDS的外显子部分。通过蛋白质保守序列选择对应外显子区域进行靶点筛选。利用(http//crispr.mit.edu/)网站分析,获得SIRT2基因sgRNA的候选序列(表1)。将SIRT2 sgRNA寡核苷酸单链退火形成双链,与BsmBⅠ酶切线性化的lentiCRISPR v2质粒连接,连接产物转化到DH5ɑ感受态中,挑取阳性单克隆菌落并提取质粒送公司测序验证。

表1 sgRNA的候选序列
3.慢病毒包装:HEK293T细胞培养于含10%胎牛血清的DMEM培养基中,细胞密度生长至60%~80%时开始转染。分别将lentiCRISPR v2质粒和lentiCRISPR-sgRNA SIRT2敲除质粒与慢病毒包装质粒pSPAX2、pMD2.G以4∶3∶1共转HEK293T细胞产生慢病毒颗粒。转染后24h更换正常培养基,48~72h收集病毒上清液,并使用离心过滤器(Merck Millipore)根据制造商的方案进行浓缩,观察其颜色和黏稠度的变化。
4.慢病毒转染及构建稳转细胞系:HEK293细胞在DMEM培养基常规培养,转染前按每孔40%的密度接种于12孔板。细胞长到80%时开始进行病毒转染。24h后用胰蛋白酶消化细胞,并以低密度重新铺板,铺板后的3h加入选择剂。用指定浓度的嘌呤霉素处理细胞。第2天更换培养基,细胞每隔1天传代1次。
5.HEK293细胞嘌呤霉素的最低致死浓度测定:将HEK293细胞接种于24孔板的12个孔,使每个孔的细胞量达到3×105,各设置两个重复。待细胞生长良好后,加入嘌呤霉素,并按0、2、4、6、8、10μg/ml浓度梯度加入每孔中。细胞培养7天后,通过观察,嘌呤霉素对HEK293细胞的最低致死浓度为2μg/ml,选择2μg/ml嘌呤霉素来抑制阴性细胞。
6.组蛋白提取:细胞用胰酶消化收集好后,向100mm培养皿中加入PBS洗涤3次,每次10ml;加入800μl含有5%蛋白酶抑制剂的Hypotonic Buffer,收集至离心管后震荡混匀,冰上放置30min后4℃离心,6000r/min,离心5min;将上清液倒掉,然后向离心管中加入0.2mol/L的硫酸600μl,充分混匀,4℃旋转过夜;离心4℃,13000r,离心min,离心5min;倒掉上清液,向离心管中加入TCA198μl,充分混匀后在冰上放置30min,每隔5min颠倒混匀1次;离心4℃,13000r/min,离心5min;弃掉上清液向离心管中加入1ml的丙酮,充分洗涤沉淀,此过程重复2~3次。离心4℃,13000r/min,离心5min;倒掉上清液后置冰上晾干,视沉淀量加入超纯水进行溶解。
7.蛋白质免疫印迹:将制备好的组蛋白样品上样,10%聚丙烯酰胺凝胶垂直电泳,电转移至PVDF膜,5%脱脂奶粉封闭后加入一定比例稀释的一抗,室温3h或4℃孵育过夜。PBST洗膜3次,每次10min,用1∶2000稀释的辣根过氧化物酶标记羊抗鼠或羊抗兔二抗室温孵育1h,PBST洗膜3次,每次10min后,用ECL化学发光试剂曝光显影,全自动化学发光成像系统(上海天能科技有限公司)扫描记录,以GAPDH作为内参,进行分析比较,实验重复3次。

结 果
1.LentiCRISPR v2-sgRNA SIRT2敲除质粒鉴定,序列测定与设计的一致:对SIRT2-sgRNA寡核苷酸单链退火形成双链,与BsmBⅠ酶切线性化的lentiCRISPR v2质粒连接,连接产物转化到DH5ɑ感受态中,通过测序证实lentiCRISPR v2-sgRNA SIRT2敲除质粒构建成功(图1)。本文构建的lentiCRISPR v2-sgRNA SIRT2敲除质粒有SIRT2-sgRNA-4序列的插入,并且序列与所设计的一致。
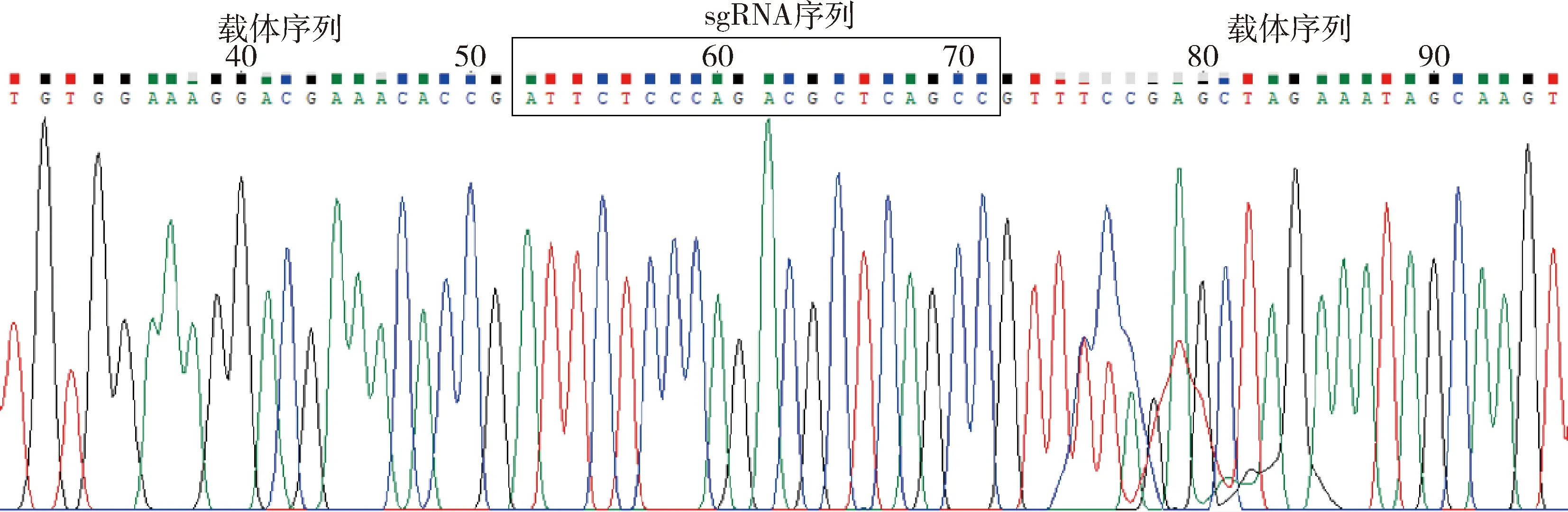
图1 构建质粒测序图
2.T7E1法验证SgRNA活性:T7E1法是验证结果的金标准。为了检测设计的sgRNA的有效性,在敲除位点的上游和下游设计PCR引物,上游引物:5′-TGTGCCTTTAGATGTGGGGA-3′;下游引物:5′-ACAAAGTGGAAGCACCTGGA-3′。长度约430bp,提取细胞基因组DNA,然后进行PCR扩增。T7E1酶可以特异性地识别由正常链和突变链所形成的杂合双链,对得到的PCR产物进行变性和退火处理后加入T7E1 (核酸内切酶,只切割碱基错配的DNA),即可检测到突变体的存在。实验结果证明,敲除(knock out, KO)了SIRT2基因的HEK293细胞其PCR扩增产物用T7E1酶切后可见明显的切割条带,说明预期的敲除位点附近存在大量sgRNA引导Cas9切割形成的缺失突变(图2)。

图2 Cas9介导的基因组中指定靶标的T7E1测定
3.SIRT2稳定敲除HEK293细胞株的构建:通过慢病毒包装并感染HEK293细胞,进一步嘌呤霉素筛选,由lentiCRISPR v2质粒包装病毒感染细胞获得的阳性克隆作为对照细胞。通过蛋白质免疫印迹验证SIRT2的敲除效率。结果显示,与对照细胞系比较,敲除细胞系中SIRT2蛋白未见明显表达,说明获得了SIRT2完全有效敲除的HEK293细胞系。利用1号和2号SIRT2稳定敲除细胞系进行后续组蛋白修饰位点的检测(图3)。
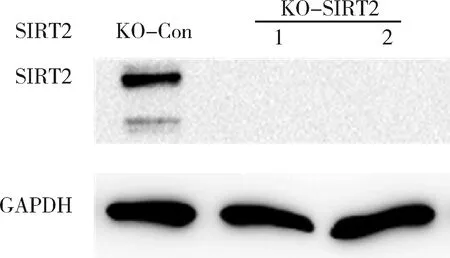
图3 与正常细胞系比较,敲除细胞系中SIRT2蛋白表达明显降低1和2为两个生物重复
4.SIRT2稳定敲除对组蛋白乙酰化修饰表达的影响:SIRT2敲除后,组蛋白H4K16乙酰化的水平与对照细胞比较显著上升,Ko-con即同等未敲除SIRT2的HEK293细胞,Ko-SIRT2即敲出了SIRT2的基因(图4)。其次检测了在SIRT2稳定敲除细胞中组蛋白H4K5乙酰化的水平。SIRT2敲除后,组蛋白H4K5乙酰化的水平与对照细胞比较下降(图5)。
5.SIRT2稳定敲除对组蛋白甲基化修饰表达的影响:进一步检测在SIRT2稳定敲除细胞中组蛋白H3K79me2甲基化的水平。SIRT2敲除后,组蛋白H3K79me2甲基化的水平与对照细胞比较升高(图6)。
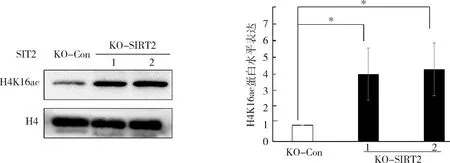
图4 SIRT2敲除以后H4K16ac的水平升高与KO-Con比较,n=3,*P<0.05;1和2为两个生物学重复

图5 SIRT2敲除以后H4K5ac的水平降低与KO-Con比较,n=3,*P<0.05;1和2为两个生物学重复

图6 SIRT2敲除以后H3K79me2的水平升高与KO-Con比较,*P<0.05;1和2为两个生物学重复
讨 论
CRISPR/Cas9系统利用慢病毒属(lentivirus) 载体将外源DNA导入细胞,该载体具有安全性高,可在体内长期表达的能力[13~16]。该系统主要是利用sgRNA引导Cas9核酸酶在靶位点处进行DNA特异性剪切,达到敲除基因的目的[1]。与其他基因编辑技术比较,该系统的构建更为简单,更能满足大多数区域的基因编辑需求。本研究利用CRISPR/Cas9系统在HEK293细胞基因组水平上成功构建了SIRT2基因敲除的细胞系。
组蛋白乙酰化和去乙酰化修饰主要有两类酶参与,即组蛋白乙酰化酶(histone acetylase,HAT)和组蛋白去乙酰化酶(histone deacetylase,HDAC)。目前有3类组蛋白的去乙酰化酶,SIRT2作为第1个被发现的Ⅲ类组蛋白的去乙酰化酶,在组蛋白的修饰中发挥重要的作用。有文献报道SIRT2可以使H4K16去乙酰化,从而在细胞分裂应激时阻止染色质凝聚。本研究首先检测了在SIRT2稳定敲除细胞中组蛋白H4K16乙酰化的水平。
SIRT2主要定位于细胞质内,同时也可以穿梭到细胞核内充当组蛋的去乙酰化酶,通过对H4K16去乙酰化作用降低G2/M期染色质的凝聚[17]。本研究发现,SIRT2稳定敲除HEK293细胞系,组蛋白H4K16乙酰化的水平与对照细胞比较显著上升,与前期文献报道一致。组蛋白H4K5乙酰化的水平与对照细胞比较下降,组蛋白H3K79me2的水平与对照细胞比较升高,说明SIRT2敲除后可能特异的调节甲基化和乙酰化的水平,但其具体调控机制尚不清楚,笔者后期还会继续探究其具体作用机制,并做进一步的研究。
SIRT2广泛分布于全身的各个组织,在代谢组织中高表达,如大脑、肝脏、脂肪等组织[18]。SIRT2通过催化不同底物的去乙酰化水平参与许多生物学过程。有研究表明,SIRT2沉默表达后,能够抑制肝癌细胞的增殖、侵袭和转移[19]。也有研究表明,在3T3-L1前脂肪细胞模型内,SIRT2与FoxO1相互作用并使其乙酰化减少,从而影响脂肪生成与脂肪细胞的分化,表明SIRT2在脂肪生成中发挥重要作用,然而SIRT2在肥胖和代谢综合征发病过程中的作用及机制尚不清楚。SIRT2是人类的Ⅲ类组蛋白去乙酰化酶,介导组蛋白及非组蛋白的去乙酰化修饰,本研究再一次验证了SIRT2稳定敲除HEK293细胞系特异的调节H4K16乙酰化的水平,该细胞系的构建为与SIRT2相关疾病的发病机制的研究提供了可能性。
本研究通过CRISPR/Cas9系统成功构建了SIRT2的稳定敲除细胞系,为进一步深入研究SIRT2对组蛋白乙酰化和甲基化的调节及其对下游靶基因的调节作用提供了依据。
猜你喜欢
中国生物化学与分子生物学报(2022年8期)2022-09-08
江西农业学报(2021年4期)2021-04-20
水生生物学报(2021年1期)2021-02-04
畜牧与饲料科学(2018年11期)2018-12-10
中国组织化学与细胞化学杂志(2016年4期)2016-02-27
医学研究杂志(2015年4期)2015-06-10
中国当代医药(2015年16期)2015-03-01
中国当代医药(2015年9期)2015-03-01
中国医药导报(2015年26期)2015-02-28
中华介入放射学电子杂志(2014年1期)2014-02-02
