
基于近红外光谱技术的砂仁提取物混合工艺研究△
2020-03-28 06:31:26刘燎原梁志毅刘丽萍钟如帆
中国现代中药 2020年1期
刘燎原,梁志毅*,刘丽萍,钟如帆
1.广东一方制药有限公司,广东 佛山 528244;2.广东省中药配方颗粒企业重点实验室,广东 佛山 528244
砂仁为姜科植物阳春砂AmomumvillosumLour.、绿壳砂AmomumvillosumLour.var.xanthioidesT.L.Wu et Senjen或海南砂AmomumlongiligulareT.L.Wu的干燥成熟果实,具有化湿开胃、温脾止泻、理气安胎的功效[1],砂仁挥发油具有抗溃疡、促进胃肠蠕动、胃肠保护等药理作用[2],为砂仁主要有效部位之一。砂仁配方颗粒在生产中需通过水蒸气蒸馏法收集挥发油,挥发油经β-环糊精包合并干燥后,与砂仁水提浸膏混合均匀后再制粒。为保证砂仁配方颗粒的临床功效,其中挥发油β-环糊精包合物混合均匀度是其重要质量指标,但砂仁挥发油类成分在β-环糊精包合物和砂仁提取物中的含量比例非常小,难以通过一般的检验手段检测提取物的混合均匀性。
传统的混合均匀度检测方法是在混合过程的不同时间点,多次暂停混合,于混合器的不同空间位置取样,通过物理、化学或仪器分析方法如高效液相色谱法、气相色谱法、紫外可见吸收光谱法等检测样品之间的相似程度和变化趋势。传统检查方法,由于样品个数多、供测定的样品制备过程繁杂、测定周期长、且消耗大量的有机试剂及对照品,检测效率较低;而近红外光谱法的定性、定量检测具有快速、非破坏性、无试剂分析、无需对照品、安全、高效、成本低及同时测定多种组分等特点[3],使其可以对混料过程中的物料均匀度进行在线检测和分析,进而应用到在线质量控制,这一技术对中药原材料的质量控制及中药制药过程的实时监控均有独特的优势。且该技术正在从传统的植物药及中药复方中个别有效成分或指标性成分的定量检测向矿物药检测、中药提取及制剂过程实时监测发展,且检测对象正在向多质量控制指标及安全性检测指标发展[4]。
本研究拟采用近红外光谱法,测定砂仁提取物中挥发油β-环糊精包合物的含量,并计算各部位的RSD,以此来判断提取物的混合均匀程度,从而优选砂仁提取物混合工艺。
1 材料
1.1 仪器
傅立叶变换近红外光谱仪(德国Bruker Tango);ME204E万分之一天平(梅特勒-托利多);HGD-8000固定料斗混合机(浙江迦南科技股份有限公司)。
1.2 试药
砂仁水提浸膏粉(批号分别为T1605011-1、T1605011-2、T1605011-3,广东一方制药有限公司);砂仁挥发油β-环糊精包合物(各批批号分别为B1605011-1、B1605011-2、B1605011-3,广东一方制药有限公司)。
2 方法
2.1 砂仁提取物混合工艺
称取砂仁水提浸膏粉约1500 kg,砂仁挥发油β-环糊精包合物约179 kg,各3份,分别将浸膏粉和挥发油β-环糊精包合物装入混合机进行混合,混合机转速设定为5 r·min-1。各批混合物中挥发油β-环糊精包合物占比见表1。
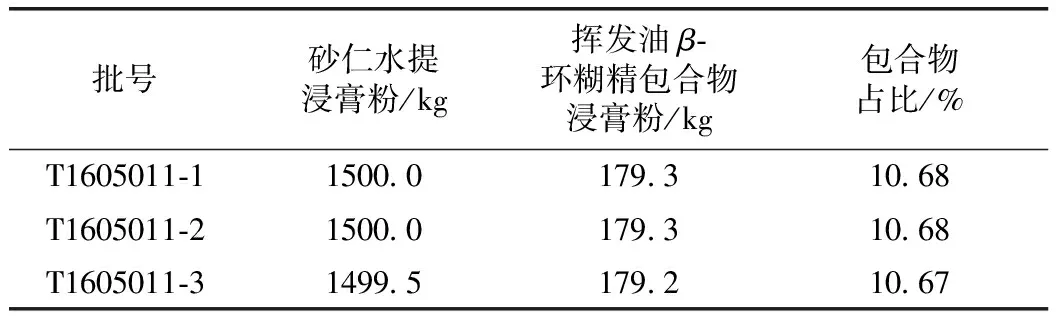
表1 各批混合物中挥发油β-环糊精包合物占比
2.2 取样方法
分别在设备开机混合运行到5、10、20、30 min时,停机,将取样器从混合机的顶部插入,在规定的取样部位进行取样,样品编号保存。将4个运行时间的样品全部取完,编号,备用。
取样部位见图1,ΔS1S4S7为等边三角形,自S1点垂直向下到接触锥体斜面处为S3点,S1与S3中间点为S2点,同样的自S4向下有取样点S5和S6,自S7向下有取样点S8和S9。样品编号与混合时间的对应关系见表2。
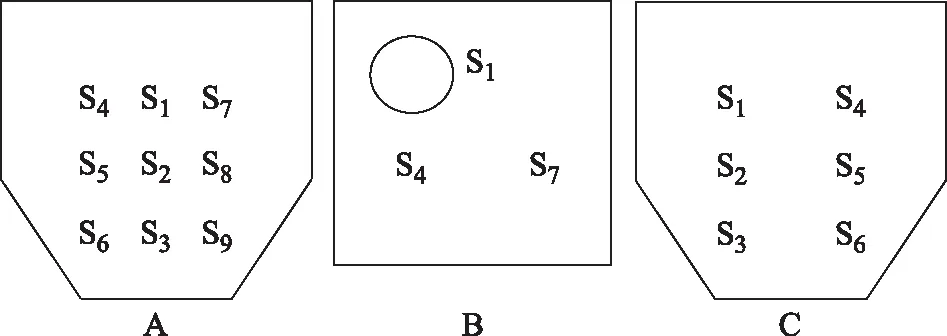
注:A.混合机取样正视图;B.混合机取样俯视图;C.混合机取样左视图。图1 混合机取样部位示意图

表2 样品编号与混合时间对应关系
2.3 砂仁挥发油β-环糊精包合物近红外模型的建立
2.3.1样品的制备 精密称取砂仁水提浸膏粉和砂仁挥发油β-环糊精包合物适量,混合均匀,并根据实际加入量计算出砂仁挥发油β-环糊精包合物所占比例,即为β-环糊精包合物含量真实值。制成样品共70份,将其分成2组,一组43份作为校正集,其挥发油β-环糊精包合物质量分数为0%~19.98%,另一组27份作为验证集,其挥发油β-环糊精包合物质量分数为3.42%~17.33%。包合物含量数据见表3。

表3 砂仁包合物含量真实值
2.3.2近红外光谱的采集 取混合均匀的砂仁提取物,置具塞玻璃样品瓶中,采用积分球漫反射,采集近红外光谱。采集条件:以仪器内置背景为参比,扫描范围4000~12 000 cm-1,分辨率8 cm-1,扫描次数32次,取平均光谱。所得砂仁提取物近红外光谱图见图2。
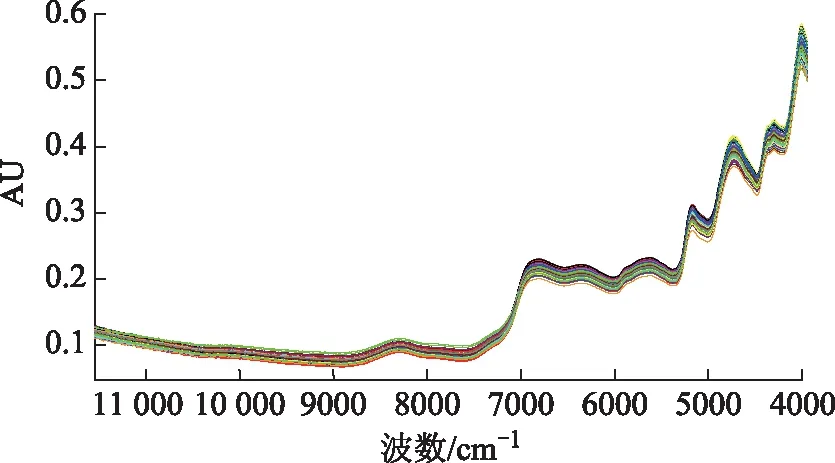
图2 砂仁提取物原始NIRS图
2.3.3模型性能评价指标 通过交叉验证的相关系数(r)、交叉验证均方差(RMSECV)、相对分析误差(RPD)和主成分维数考察模型性能,选择合适的光谱范围及光谱预处理方法建立定量模型。以r无限趋近于1,RMSECV越小,评价模型性能越好[5]。采用近红外OPUS7.5分析软件,自动优选预处理方法与光谱范围,优化结果见表4,预处理后的光谱图见图3。方法1“一阶导数+矢量归一化”和方法2“一阶导数+MSC”2种处理方法的RMSECV、RPD、光谱范围、r及维数结果均相同,故选择其中1种处理方法即可。采用“一阶导数+MSC”法对原始吸收光谱进行预处理,建模的光谱范围选取7 500.1~5 447.6 cm-1特征波段,维数选为3,经交叉验证r=0.995 5,RMSECV=0.383,RPD=14.9。
2.3.4近红外定量模型的建立 采用近红外OPUS7.5分析软件,选择2.3.3项下的预处理方法与光谱范围。运用偏最小二乘(PLS)法建立砂仁挥发油β-环糊精包合物定量校正模型,其预测值与真实值相关图见图4。

表4 砂仁挥发油β-环糊精包合物模型在不同光谱范围与预处理方法下的模型参数
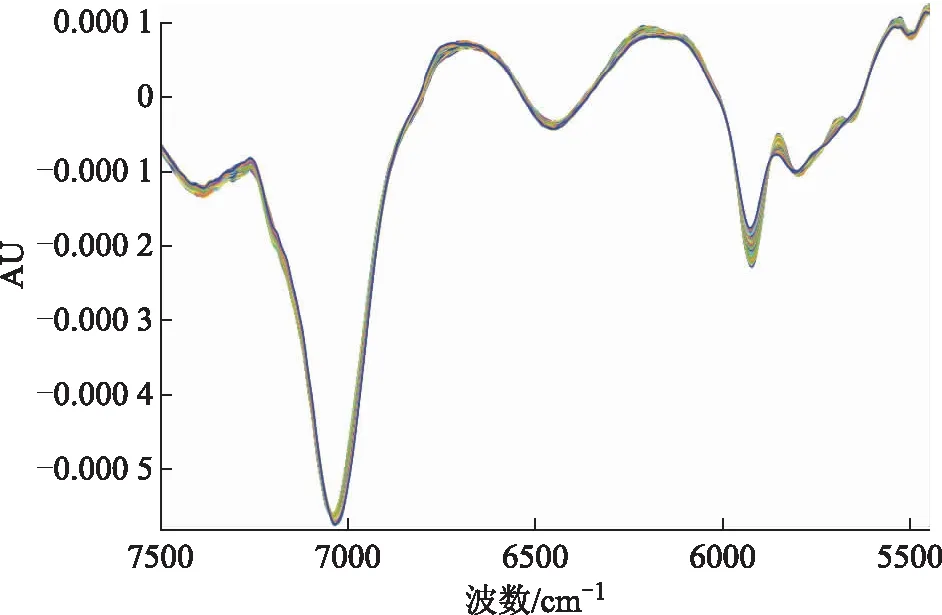
图3 砂仁提取物“一阶导数+MSC”处理图
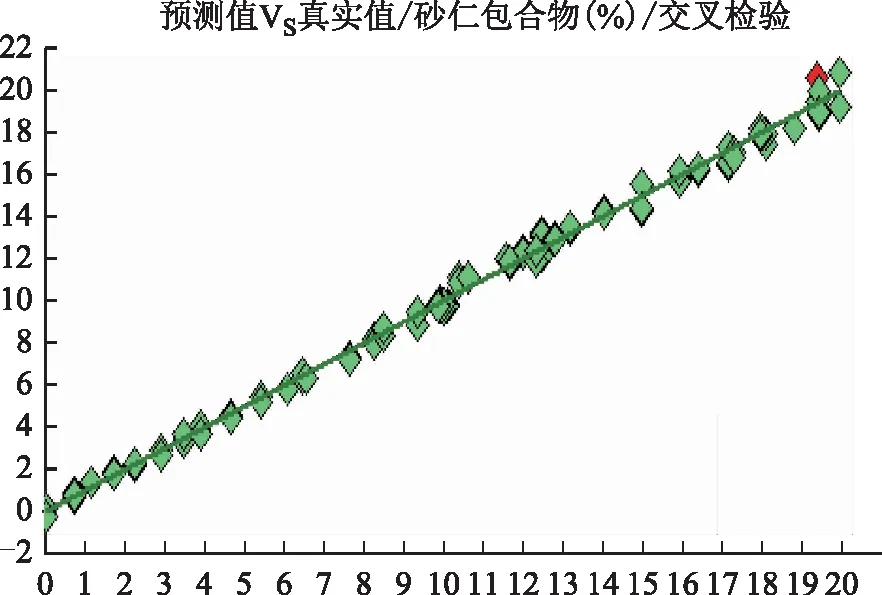
注:维数为3;r=0.995 5;RMSECV=0.383;偏移为-0.002 06;RPD为14.9。图4 挥发油β-环糊精包合物定量校正模型预测值与真实值相关图
2.3.5近红外定量模型的验证 将27个验证集样品近红外图谱导入2.3.4项下所建砂仁挥发油β-环糊精包合物定量模型,预测其砂仁挥发油β-环糊精包合物的含量,并与真实值进行比较,平均相对偏差为0.98%,平均预测回收率为98.22%,在可接受范围内,表明该方法可行。
2.3.6方法学考察
2.3.6.1精密度试验 取同一供试品(T1605011-3-C8)重复扫描,按2.3.4项下的方法测量8次,所得砂仁挥发油β-环糊精包合物含量预测值的RSD为2.67%,表明该方法精密度良好。
2.3.6.2重复性试验 取同一批号供试品(T1605011-3-C8),分别取6 份样品,按2.3.4项下的方法采集光谱,所得砂仁挥发油β-环糊精包合物含量预测值的RSD值为2.34%,表明该方法重复性良好。
2.3.6.3稳定性试验 取同一供试品(T1605011-3-C8),分别于取样后0、1、2、3、4、5、6 h,按2.3.4项下的方法采集光谱,所得砂仁挥发油β-环糊精包合物含量预测值的RSD为2.70%,表明该方法稳定性良好。
2.4 砂仁提取物中挥发油β-环糊精包合物含量测定结果
按2.2项下方法,取砂仁提取物(批号分别为T1605011-1、T1605011-2、T1605011-3,每批各36份)供试品共108份,按2.3.4项下的方法扫描测量,结果见表5。
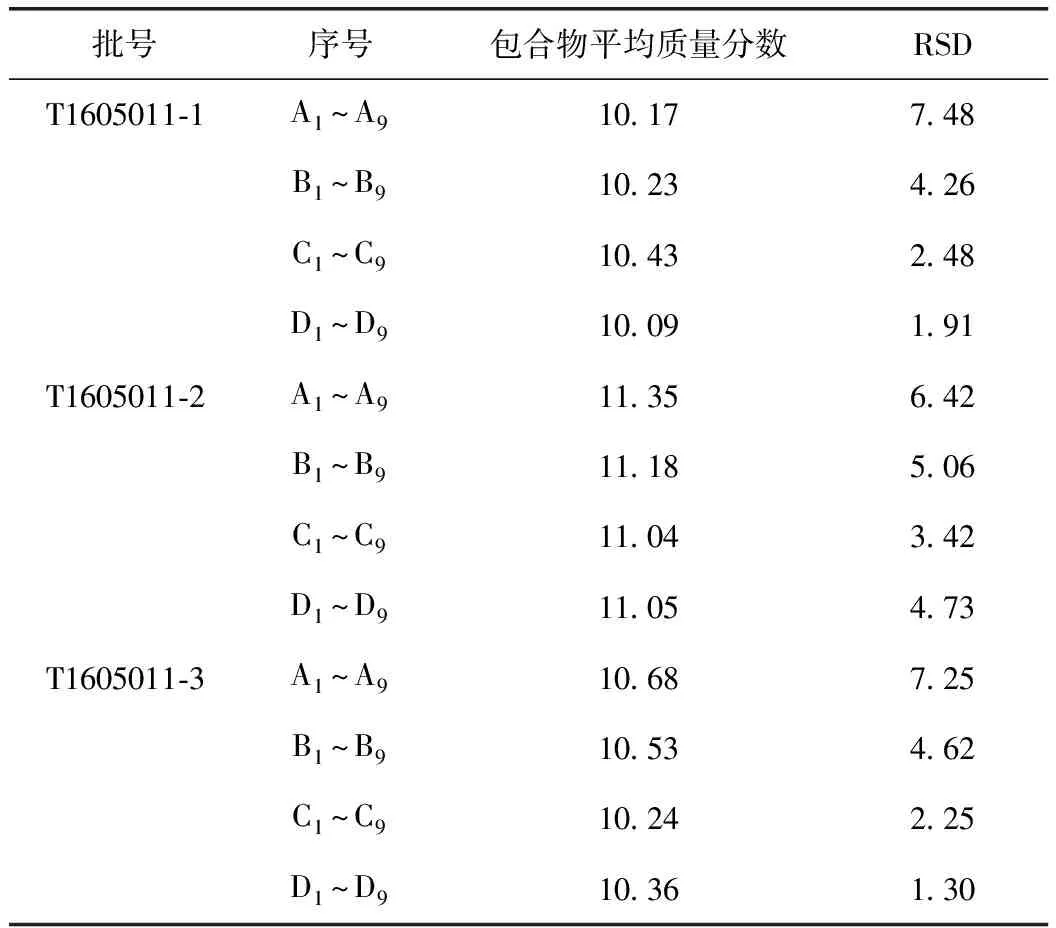
表5 砂仁提取物中挥发油β-环糊精包合物含量测定结果 %
由表可知,砂仁提取物中挥发油β-环糊精包合物的含量测定结果显示,3批样品在混合20 min以后,提取物不同部位中挥发油β-环糊精包合物含量测定的RSD分别为2.48%、3.42%、2.25%,均小于5.0%,说明混合均匀,确定砂仁提取物混合工艺为:混合转速5 r·min-1,混合时间20 min。
3 讨论
本研究通过建立砂仁提取物近红外含量检测模型并进行方法学考察,建立了近红外光谱快速测定砂仁提取物中挥发油β-环糊精包合物含量检测方法,解决了采用高效液相色谱法、气相色谱法检测样品制备过程繁杂、测定周期长、消耗大量的有机试剂及对照品、效率较低的问题,提高了测定效率。
应用近红外光谱技术建立砂仁提取物中挥发油β-环糊精包合物的定量测定模型,测定了混合不同时间砂仁提取物中挥发油β-环糊精包合物的含量,确
定了砂仁提取物的混合工艺条件,确保最终产品中各组分分布的均匀性,实现药品均一性,同时可实现砂仁水提浸膏粉与挥发油包合物混合的在线均匀度检测,提高药品质量的可控性。
随着分析仪器的不断改进、计算机的快速发展以及数据处理方法的应用,近红外光谱凭借其独特的优势,发展成为了一门独立的分析技术,广泛应用于各个领域。近红外光谱分析技术在制药过程主要环节也均有应用研究,包括原料药的质量控制、化学反应过程监测、制剂过程监测等[6-8]。而其在中药配方颗粒领域应用还鲜有报道,本研究将近红外光谱分析技术应用于配方颗粒制备过程中混合工艺的质量控制,为今后近红外光谱技术在配方颗粒领域的应用提供参考,对提高配方颗粒生产以及质量控制效率具有重要的现实意义。
猜你喜欢
保健与生活(2019年5期)2019-08-01 06:31:38
中成药(2018年8期)2018-08-29 01:28:08
中成药(2018年5期)2018-06-06 03:11:49
中成药(2018年4期)2018-04-26 07:12:43
中成药(2017年12期)2018-01-19 02:06:56
源流(2017年9期)2017-10-29 08:57:52
中成药(2017年5期)2017-06-13 13:01:12
益寿宝典(2017年36期)2017-02-26 06:10:06
中国中医药现代远程教育(2014年15期)2014-03-01 04:28:11
食品科学(2013年13期)2013-03-11 18:24:19
