
低温微萃取-GCMS/MS联用技术检测食用油中20种持久性污染物
2020-03-28 04:12:26刘笑笑张菁菁李晨曦吴福祥姬良亮丁辉彭涛
食品与发酵工业 2020年5期
刘笑笑,张菁菁, 李晨曦,吴福祥,姬良亮,丁辉, 彭涛
(兰州市食品药品检验所, 甘肃 兰州,740050)
多环芳烃(polycylic aromatic hydrocarbon, PAHs)和塑化剂(plasticizer PAEs)属于持久性环境有机污染物,具有高毒性、易迁移性、稳定性和生物累积性等特点[1-2],大量存在于水、土壤、蔬菜以及动物组织中。其化学性质稳定且很难被生物降解[3],易被人体吸收产生巨大危害,可通过食物链放大逐级传递[4]。膳食暴露是PAEs,PAHs这些持久性污染物进入人体的主要途径[5-7],食品中持久性污染物残留问题已受到国际社会广泛的关注。食用油是这些持久性污染物PAHs,PAEs通过饮食进入人体的主要途径之一[8-11], PAHs,PAEs含量水平成为评价食用油质量安全的重要指标[12-16]。因此,研究PAE及PAH的高效检测方法,对更为全面地监测和评价食用油污染状况[17-19],保证食用油质量安全具有重要意义。
目前PAHs较多采用液相色谱法(HPLC)和气相质谱法(GC-MS)[20],该方法时间较长,干扰较大。PAEs较多采用GC-MS法,该方法对部分复杂样品干扰较大,易出现假阳性结果,检验标准相对单一、低效、滞后[21-22]。相较于传统方法,GC-MS/MS具有较高灵敏度和分辨率,能够更有效地消除其他杂质的干扰,提高离子选择性和准确性[12],并降低了检测限度。多反应监测模式(multiple reaction monitoring, MRM)为食用油中微量持久性污染物的检测提供新的有效手段[23]。由于食用油样品基质干扰复杂,建立快速有效检测植物油中持久性污染物PAEs和PAHs的技术很有必要。本研究使用低温除脂微萃取技术,结合GC-MS/MS的高灵敏、高选择性,建立了检测食用植物油中PAHs和PAEs的新方法,并进行了实际应用。
1 材料与方法
1.1 材料与仪器
1.1.1 材料
标准物质:6种PAEs对照品,Dr.Ehrenstorfer GmbH公司:邻苯二甲酸二甲酯(dimethyl phthalate,DMP)99.5%、邻苯二甲酸二乙酯(diethyl phthalate,DEP)99.5%、邻苯二甲酸二异丁酯(diisobutyl phthalate,DIBP)99.5%、邻苯二甲酸二丁酯(dibutyl phthalate,DBP)99.4%、邻苯二甲酸二戊酯(di-N-pentyl phthalate,DPP)99.0%、邻苯二甲酸二己酯(di-n-hexyl phthalate,DHXP)99.5%。14种PAHs标准品,Sigma公司: 萘(naphthalene,NAP)(2 000±20.39)mg/L,99.9%;苊烯(acenaphthylene,ANY)(2 000±7.22) mg/L,99.7%;蒽(anthracene,ANT)(2 004±20.44)mg/L,99.2%;荧蒽(fluoranthene,FLT)(1 999±4.48) mg/L,99.2%;菲(phenanthrene,PHE)(2 000±4.48)mg/L,99.5%;芴(fluorene,FLU)(2 003±10.79)mg/L,99%;苊(acenaphthene,ANA)(2 001±10.78) mg/L,99%;芘(pyrene,PYR)(2 001±4.49) mg/L,98.4%;苯并(a)蒽(benzo(a)anthracene,BaA)(2 002±20.42) mg/L,99%;(chrysene,CHR)(2 002±20.42) mg/L,98%;苯并(b)荧蒽(benzo[b] fluoranthene,BbFA)(2 000±4.48)mg/L,99.8%;苯并(k)荧蒽(benzo[k] fluoranthene,BkFA)(2 000±4.48)mg/L,99.3%;苯并(a)芘(benzo[a] pyrene,BaP)(1 998±4.48) mg/L,99.5%,二苯并(h)蒽(dibenz[a,h] anthracene,DhA)(2 001±10.78) mg/L,99.5%。
试剂:乙腈、正己烷、石油醚、甲醇、二氯甲烷(色谱纯),默克公司;8000D气相色谱-质谱仪,安捷伦公司;ME403分析天平,梅特勒托莱多公司;GENIUS3涡旋混合仪,德国IKA公司;GVA50A氮吹仪,北京普立泰科仪器有限公司;0.1~10 mL移液枪,德国Eppendorf公司;B-400均质仪,瑞士BUCHI公司。
1.1.2 仪器
1.2 实验方法
1.2.1 样品前处理
准确称取1.0 g食用油样品,置于20 mL比色管中, 丙酮作为分散剂,加入10 mLV(二氯甲烷)∶V(丙酮)=1∶1溶剂,于涡旋混合器上旋涡提取(2 000 r/min)0.5 min,再超声萃取8 min,10 000 r/min离心5 min,使溶液充分分层后,置于-20 ℃冷冻除脂60 min,将上层清液倒入氮吹管中,低温氮吹至干,准确加入二氯甲烷2 mL,涡旋5 s,转移至进样瓶中,上机检测。试剂空白样,除不加样品外,其他操作相同。
1.2.2 气相色谱-质谱检测
1.2.2.1 色谱条件
色谱柱:DB-5MS 毛细管柱(Agilent19091S-433UI,30 m×250 μm×0.25 μm);载气纯度高于99.999%的高纯氮气,流速1.0 mL/min;进样口温度250 ℃,不分流进样,进样体积为1μL;升温程序:初始柱温70 ℃,保持2 min,以 25 ℃/min 升温至150 ℃,保持1 min, 再以3 ℃/min 升温至200 ℃,保持1 min,以8 ℃/min 升温至280 ℃,保持10 min。
1.2.2.2 质谱条件
电子轰击离子源:电离能量70 eV,离子源温度250 ℃,传输线温度280 ℃,溶剂延迟5 min,灯丝电流100 μA,MRM多反应检测模式,其具体参数见表1。
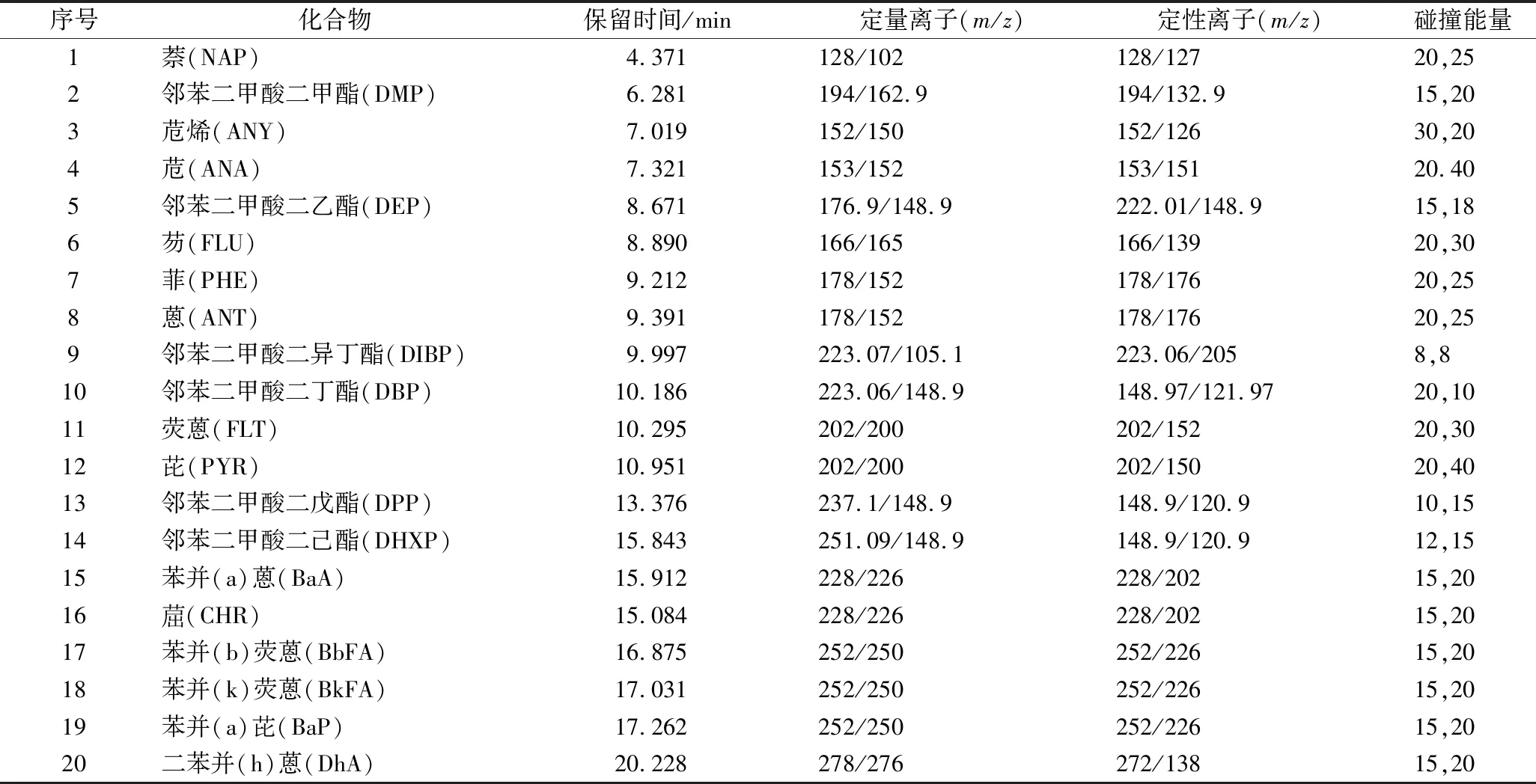
表1 多重反应监测参数
2 结果与分析
2.1 前处理方法的建立
2.1.1 分散剂的选择
考察了丙酮、乙腈、乙醇和甲醇4种分散剂对目标物萃取效率的影响。结果表明,除了萘在乙醇作分散剂时萃取效率略微比丙酮高之外,其余19种污染物均在丙酮做分散剂时萃取效率最高,故选择丙酮作为分散剂。
2.1.2 萃取剂的选择
本文选用10 μg/L混合标准溶液进行加标实验,分别选用3种不同体系V(二氯甲烷)∶V(丙酮)=1∶1、V(正己烷)∶V(丙酮)=1∶1、V(乙腈)∶V(丙酮)=1∶1多环芳烃萃取剂进行加速溶剂萃取试验,如图1所示,不同提取液提取效率不尽相同,总体呈现出二环、五环和六环的PAHs提取效率较低,三环和四环的 PAHs提取效率较高。其中,二氯甲烷-丙酮萃取平均回收率是82.1%~101%,对20种污染物均能达到较为满意的萃取率,正己烷-丙酮和乙腈-丙酮萃取平均回收率分别为64.5%~88.3%、53.8%~85.6%,DMP、ANY、DEP、BkFA和BaP的回收率较低,平均萃取效果不如二氯甲烷-丙酮,综合考虑各个目标物定量的准确性,试验确定V(二氯甲烷)∶V(丙酮)=1∶1混合溶剂(下简称二氯甲烷-丙酮溶剂)作为样品的萃取剂。

1-NAP;2-DMP;3-ANY;4-ANA;5-DEP;6-FLU;7-PHE;8-ANT;9-DIBP;10-DBP;11-FLT;12-PYR;13-DPP;14-DHXP;15-BaA;16-CHR;17-BbFA;18-BkFA;19-BaP;20-DhA
2.1.3 萃取方式的选择
称取1 g待测样,加入2、5、10、12、15 mL二氯甲烷-丙酮萃取溶剂,涡旋分散30 s,再超声1、2、5、8、15 min后提取,考察萃取剂体积和超声萃取时间对萃取率的影响,结果如图2所示,萃取溶剂加入量10 mL,涡旋分散30 s,再超声萃取8 min为最佳萃取方式。

图2 不同萃取方式对提取效果的影响
2.1.4 冷冻时间的优化
称取1 g待测样,加入10 mL二氯甲烷-丙酮溶剂,涡旋分散30 s再超声萃取8 min后,分别选择温度为-4、-10、-15、-20、-25 ℃,分别放置10、30、60、90、120、150、200 min冷冻除脂,考察冷冻温度和冷冻时间对除脂净化的影响,结果如图3所示,-20 ℃冷冻除脂60 min为最佳冷冻条件。冷冻后,上层提取液可方便地倾入鸡心瓶中,实现与油脂充分分离。

图3 冷冻脱脂时间的优化
2.1.5 反萃取剂的选择及用量
称取1 g待测样,加入10 mL二氯甲烷-丙酮溶剂,涡旋分散30 s再超声萃取8 min,高速离心等溶液分层后,置于-20 ℃冷冻除脂60 min,考虑反萃取效果,将低温脱脂的溶剂蒸干,选择40°水浴,旋转蒸发时,当旋蒸至近干时, NAP损失较为严重, ANY、ANA也有一定的损失,因此,选用低温氮气吹干,可有效避免上述损失。选择常用的乙腈、甲醇、二氯甲烷、正己烷作为反萃取剂,加入量分别为0.5、1.0、1.5、2.0、2.5、5.0 mL,比较其回收率,结果如图4所示,最终选择用2 mL二氯甲烷对其进行反萃取。
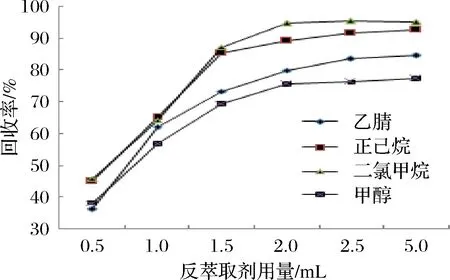
图4 反萃取剂的选择
2.2 GC-MS/MS检测技术的建立
2.2.1 质谱条件的优化
以全扫的方式对20种目标物进行扫描,获得其总离子流图,根据质谱图,选择分子量较大,丰度较高且干扰较少的碎片作为特征离子,见图5。

1-NAP;2-DMP;3-ANY;4-ANA;5-DEP;6-FLU;7-PHE;8-ANT;9-DIBP;10-DBP;11-FLT;12-PYR;13-DPP;14-DHXP;15-BaA;16-CHR;17-BbFA;18-BkFA;19-BaP;20-DhA
2.2.2 线性范围与检出限
由于持久性污染物在大自然界中普遍存在且不容易降解,在空白溶液中均有检出,本实验采取扣除空白样品的方法,以消除试剂本底对检测结果的影响,实验发现,样品基质对目标分析物的基质效应既有增强也有抑制,因此采取基质配标来降低基质效应的影响,以提高定性、定量的准确度,结果见表2。
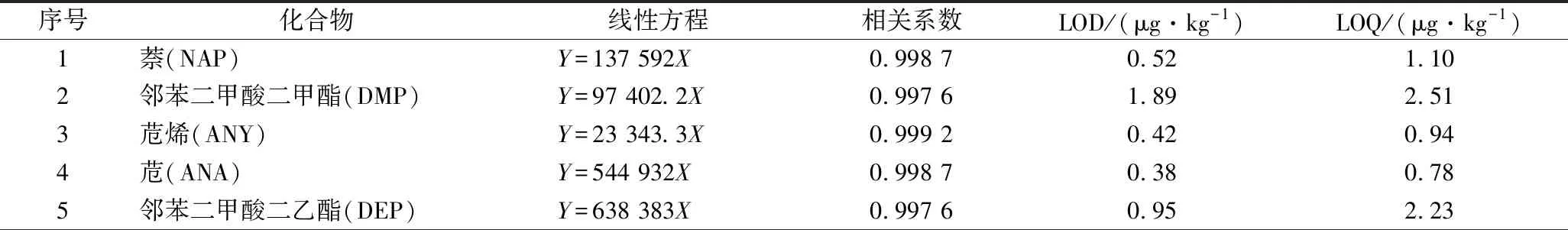
表2 植物油中20种PAEs和PAHs的线性方程、相关系数、检出限及定量限
续表2
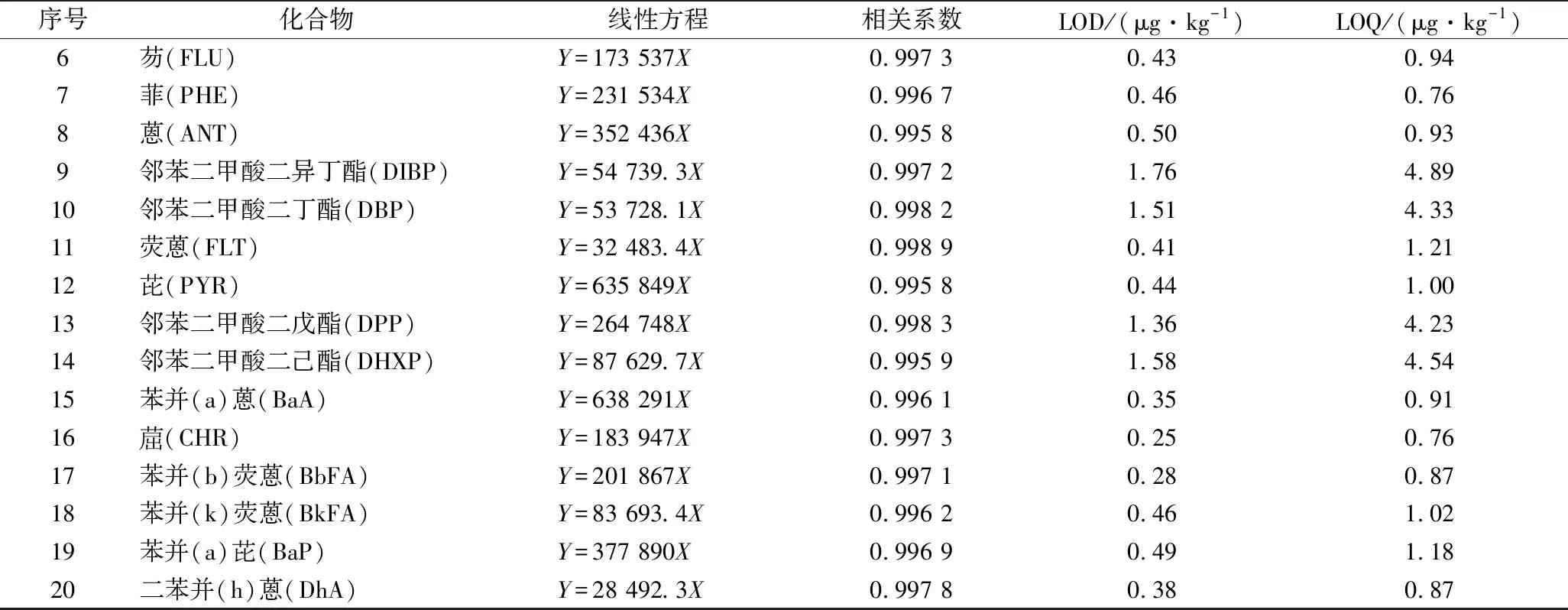
序号化合物线性方程相关系数LOD/(μg·kg-1)LOQ/(μg·kg-1)6芴(FLU)Y=173 537X0.997 30.430.947菲(PHE)Y=231 534X0.996 70.460.768蒽(ANT)Y=352 436X0.995 80.500.939邻苯二甲酸二异丁酯(DIBP)Y=54 739.3X0.997 21.764.8910邻苯二甲酸二丁酯(DBP)Y=53 728.1X0.998 21.514.3311荧蒽(FLT)Y=32 483.4X0.998 90.411.2112芘(PYR)Y=635 849X0.995 80.441.0013邻苯二甲酸二戊酯(DPP)Y=264 748X0.998 31.364.2314邻苯二甲酸二己酯(DHXP)Y=87 629.7X0.995 91.584.5415苯并(a)蒽(BaA)Y=638 291X0.996 10.350.9116(CHR)Y=183 947X0.997 30.250.7617苯并(b)荧蒽(BbFA)Y=201 867X0.997 10.280.8718苯并(k)荧蒽(BkFA)Y=83 693.4X0.996 20.461.0219苯并(a)芘(BaP)Y=377 890X0.996 90.491.1820二苯并(h)蒽(DhA)Y=28 492.3X0.997 80.380.87
2.2.3 提取净化方法的验证
选取空白的植物油样本,分别添加10、20、30 μg/kg的标准混合溶液,按照前处理方法进行加标回收和精密度试验,结果如表3所示:平均回收率为51.34%~116.2%,相对标准偏差(relative standard deviation, RSD)为0.9%~12.1%。结果表明,20种待测化合物加标回收率均在70%~120%。

表3 20种多环芳烃和增塑剂的回收率与相对标准偏差(n=5) 单位:%
2.4 食用油实际样品检测
应用本方法检测了市售8种食用油,每个样品重复检测2次,从表4可以看出,不同种类、不同生产日期、不同包装的食用油都检出PAEs和PAHs。8个油样中全部检出DBP和BaP,其含量分别在3.3~106.7 μg/kg和0.4~5.6 μg/kg。6个油样中检出DMP,其含量为39.3~265.1 μg/kg。所有油样中没有检测出ANY,对照GB 2762—2017《食品安全国家标准食品中污染物限量》油脂中BaP规定限量为5 μg/kg,GB 9685—2008 中规定DBP≤0.3 mg/kg,本实验所测定的8种食用油中BaP超标率12.5%,DBP超标率为0,其他能检测出的PAEs和PAHs在国家限量标准中并未列出,而欧盟委员会835/2011号文件规定油脂中BaP的最高残留限量值为2 μg/kg,4种PAHs(BaA、CHR、BFA、BaP)之和的限量为10 μg/kg。欧盟“食品接触塑料材料和制品法规”中规定了DBP含量≤0.05%。几乎每种食用油中均有不同程度的PAEs和PAHs检出,这可能与食用油包装材质中污染物迁移有关,也可能与油脂生产中所用加工助剂以及设备和管件材质有关。

表4 不同食用油中塑化剂和多环芳烃含量的检测结果 单位:μg/kg
注:ND为未检出,NQ为定量限
3 结论
建立了快速测定植物油中6种邻苯二甲酸酯(PAEs)和14种多环芳烃(PAHs)的气相色谱-串联质谱(GC-MS/MS)分析方法。20种目标物在5~200 μg/L范围内呈良好线性,线性相关系数均大于0.995,方法定量下限为1.0 μg/kg。在10、20、30 μg/kg 3个加标水平下的平均回收率为51.34%~116.2%,相对标准偏差为0.9%~12.1%。说明该技术适用于20种PAHs和PAHs的检验前处理,方法简便、快捷、准确,可用于植物油中增塑剂和多环芳烃残留的检测,为植物油的质量控制和安全评价提供了保证。对实际样品进行检测,均检出PAEs和PAHs,对照国内限量BaP超标率12.5%。不合格率远低于欧盟标准,国家限量标准有待完善。
猜你喜欢
食品安全导刊(2021年21期)2021-08-30 08:21:48
化工管理(2021年7期)2021-05-13 00:44:56
新疆大学学报(自然科学版)(中英文)(2020年2期)2020-07-25 01:40:26
上海化工(2018年10期)2018-10-31 01:21:06
上海农业学报(2017年3期)2017-04-10 12:39:20
淮南师范学院学报(2015年3期)2015-03-22 01:16:14
中华皮肤科杂志(2014年3期)2014-12-19 12:54:56
中国药理学通报(2014年2期)2014-05-09 08:22:20
同位素(2014年2期)2014-04-16 04:57:13
化工生产与技术(2014年5期)2014-02-27 13:42:02
