
超高效液相色谱-四极杆-飞行时间质谱快速筛查中成药、保健品中26种非法添加化合物
2020-03-28 04:12王雨晴王雨辰陈亮周明王越群谷瑞增冀峰郑锌
食品与发酵工业 2020年5期
王雨晴,王雨辰,陈亮,周明,王越群,谷瑞增,冀峰,郑锌*
1(中国食品发酵工业研究院有限公司,北京市蛋白功能肽工程技术研究中心,北京,100015) 2(岛津企业管理(中国)有限公司,北京,100020)
对保健食品中非法添加物的检测,已经从单一化合物检测,逐步发展为多种功效成分同时定性定量分析,并且涉及的非法添加化合物的品种呈递增趋势。与低分辨的三重四极杆质谱不同,高分辨飞行时间质谱能够获得高分辨率的一级质谱信息,以及特征的二级质谱图,目标化合物数量不受限制。利用二级质谱信息建立私人数据库,扩展性强,无需标准品即可实现对多种未知非法添加物的筛查。此外,高分辨质谱数据还可进行回溯研究,实现样品一针进样,样品信息全部记录,后期可依据不同数据库筛选更多目标物质。
常见的化学添加物主要有二甲双胍类降糖药、胰岛素增敏剂、那非类药物等[1-3]。国家市场监督管理总局(原食品药品监督管理局)发布的《保健食品中75种非法添加化学药物的检测》[2017年第138号][4]、《食品中那非类物质的测定》[2018年第14号][5]等相关标准及补充检验方法,覆盖了市面上主要的非法添加药物种类。对于中成药、保健品中的非法添加药物的定性检测,已有相关的文献报道[6-9]。液相色谱-串联质谱法是一种主流的检测手段[10-11],此外,液相色谱法、核磁[12-13]、气质联用法[14]等也被广泛用于非法添加物的定性分析。但是,以上方法需要大量的标准品、或者制备出纯度较高的样品,分析成本高、研究延续性不强,无法应对日益增加的非法添加品种。也有研究采用液相色谱-高分辨质谱法对保健品中的非法添加物进行分析[15-19]。于泓等[19]建立了食品中多种那非类化合物的高分辨质谱定性方法,但灵敏度低、没有建立二级质谱库,方法通用性不强。依据未知峰的高分辨质荷比及同位素丰度比,可获得化合物元素组成信息。对于未知化合物结构信息,可根据其二级质谱进一步判断或进行数据库检索。常见的公共数据库,如Chemspider,包含多达7 000余种化合物的分子式、结构等信息,分子式检索结果多,命中率低,无法借助二级质谱信息缩小筛选范围。因此,自建二级质谱图库,可有效应对同类化合物、同样前处理方式的指定范围的筛查,尤其适用于非法添加的定性分析。
本文基于超高效液相色谱-四极杆飞行时间质谱技术,建立了一种高通量定性检测26种降糖或抗疲劳药物非法添加物的方法,创建了相应的二级质谱库。并将该方法应用于保健品的快速筛查,方法学指标满足相关法规要求。
1 材料与方法
1.1 材料与试剂
甲醇、乙腈,均为色谱纯,德国MERCK公司;甲酸(色谱纯),天津Dikma公司;乙酸铵(色谱纯),美国Sigma-Aldrich公司;超纯水(18.2 MΩ·cm),法国Millipore制水机制备。
盐酸丁二胍、盐酸苯乙双胍、马来酸罗格列酮、盐酸吡格列酮、格列吡嗪、甲苯磺丁脲、瑞格列奈、格列齐特、格列波脲、格列本脲、格列美脲、格列喹酮,美国Sigma-Aldrich公司,纯度均>95%;去甲基卡波地那非、盐酸伐地那非、那红地那非、红地那非、枸橼酸羟基豪莫西地那非、枸橼酸西地那非、豪莫西地那非、丙氧基艾地那非、氨基他达那非、羟丙基去甲基他达那非、N-乙基他达那非、伪伐地那非、那末西地那非、N-去甲基西地那非,德国Dr. Ehrenstorfer公司,纯度均>95%。
1.2 仪器与设备
Nexera超高效液相色谱仪系统—LCMS-9030四极杆飞行时间质谱仪(配备ESI电离源和LabSolutions Ver 5.96色谱工作站),日本岛津公司;涡旋混合仪,美国VORTEX GENIE公司;Beckman 高速冷冻离心机,美国贝克曼公司。
1.3 方法
1.3.1 标准溶液的配制
分别称取26种对照品约10.0 mg,用甲醇定容至10 mL,配制成0.289 9~1.254 6 mg/mL的单标贮备液。以乙腈为溶剂,将26种单标贮备液稀释为1 000倍,即得0.289 9~1.254 6 mg/L的混合标准工作溶液。将上述混合标准工作液用乙腈逐级稀释,系列标准工作溶液浓度分别为L1(0.002 3~0.009 8 mg/L)、L2(0.004 5~0.019 6 mg/L)、L3(0.009 1~0.039 2 mg/L)、L4(0.018 1~0.078 4 mg/L)、L5(0.036 2~0.156 8 mg/L)、L6(0.072 5~0.313 7 mg/L)、L7(0.145 0~0.627 3 mg/L)和L8(0.289 9~1.254 6 mg/L)。
1.3.2 前处理方法
称取0.1 g保健品(片剂研磨粉碎),用1 mL超纯水溶解,振摇1 min,静置10 min,加入4 mL乙腈,超声10 min,12 000 r/min离心5 min,取1 mL上清液,0.22 μm聚四氟乙烯滤膜过滤后进样分析。
1.3.3 液相色谱—质谱条件
色谱条件:流动相:A相为体积分数0.1%甲酸水溶液,B相为体积分数0.1%甲酸乙腈溶液;流速0.4 mL/min;色谱柱:Phenomenex Kinetex C18(2.1 mm I.D. ×100 mm L, 2.6 μm);柱温40℃;洗脱方式为梯度洗脱,B相初始浓度为10%(0~0.3 min),40%~95%(4.0~5.5 min),95%(7.5 min),10%(7.6~10 min),进样体积2 μL。
质谱条件:离子源:ESI(+);接口电压4.5 kV;雾化气3.0 L/min;加热气流量10.0 L/min;干燥气流量10.0 L/min;接口温度300 ℃;DL管温度250 ℃;加热块温度400 ℃;校准方法:外标法校准质量数(调谐液为NaI,质量浓度400 mg/L),分辨率>30 000,质量误差<2×10-6;扫描模式:MS全扫描m/z100~600、MS/MS(DDA)采集m/z50~600。
1.3.4 线性范围及检出限、定量限的确定
对系列标准工作溶液进行分析,以一级提取离子流色谱图峰面积(Y)对浓度(X)做标准曲线,并使用LabSolutions软件进行线性拟合,记录标准曲线的方程及线性相关系数。以信噪比S/N=3、S/N=10分别计算方法的检出限、定量限。依据测定结果确定线性范围。
1.3.5 准确度和精密度试验
以标准曲线浓度L6(0.072 5~0.313 7 mg/L)点连续分析6次,考察仪器条件的重复性,并记录保留时间及峰面积的相对标准偏差(relative standard deviations, RSD)。
以阿卡波糖片为空白样品,添加适量的单标贮备液,配制成浓度与标准曲线L5(0.036 2~0.156 8 mg/L)和L7(0.145 0~0.627 3 mg/L)相同的加标样品,按照1.3.2制备,每个样品平行6份。标准曲线法计算空白添加样品的浓度,并以此计算加标回收率及精密度。
1.3.6 数据库的建立
在上述色谱分离条件下对26种非法添加化合物标准溶液进行分析,获得26种化合物的保留时间、精确分子质量以及离子化方式。采集不同碰撞能量(10、20、30、40和50 V)下的目标化合物的二级质谱图,在ACD Labs/2012软件中输入每种化合物的名称、分子式、CAS号、精确相对分子质量和保留时间以及二级质谱图,建立二级质谱库,用于未知物二级质谱图相似度评分。
1.3.7 方法的实际应用
选用市售3份保健品或中成药,其样品前处理和检测方法参照1.3.2和1.3.3,以此评价该方法的实际应用效果。
利用本文建立的前处理和色谱、质谱条件,对3份市售的保健品及中成药(保健品2份,中成药1份)进行分析。
2 结果与分析
2.1 定性分析
根据待测化合物的结构特征,选择正离子模式进行质谱参数的优化。格列苯脲等26种非法添加化合物均能形成[M+H]+准分子离子峰。26种化合物的分子式、精确质荷比等信息如表1所示,其中伪伐地那非与那末西地那非互为同分异构体,经过单标确认,伪伐地那非、那末西地那非保留时间分别为5.52、5.98 min,可实现色谱基线分离。盐酸伐地那非在溶液中呈现离子状态,脱掉了盐酸基团,与豪莫西地那非互为同分异构体,一级精确质荷比均为489.227 85,伐地那非和豪莫西地那非保留时间分别为3.46 min和4.05 min,实现了完全的基线分离。通过精确分子量软件计算26种目标化合物的理论[M+H]+峰,实测质荷比与理论值的偏差均小于2×10-6,优于定性的一般要求(<5×10-6)。26种目标化合物的一级提取离子流图(extracted ion chromatogram,XIC)如图1所示,26种降糖或抗疲劳类化合物分离度良好,通道无干扰。
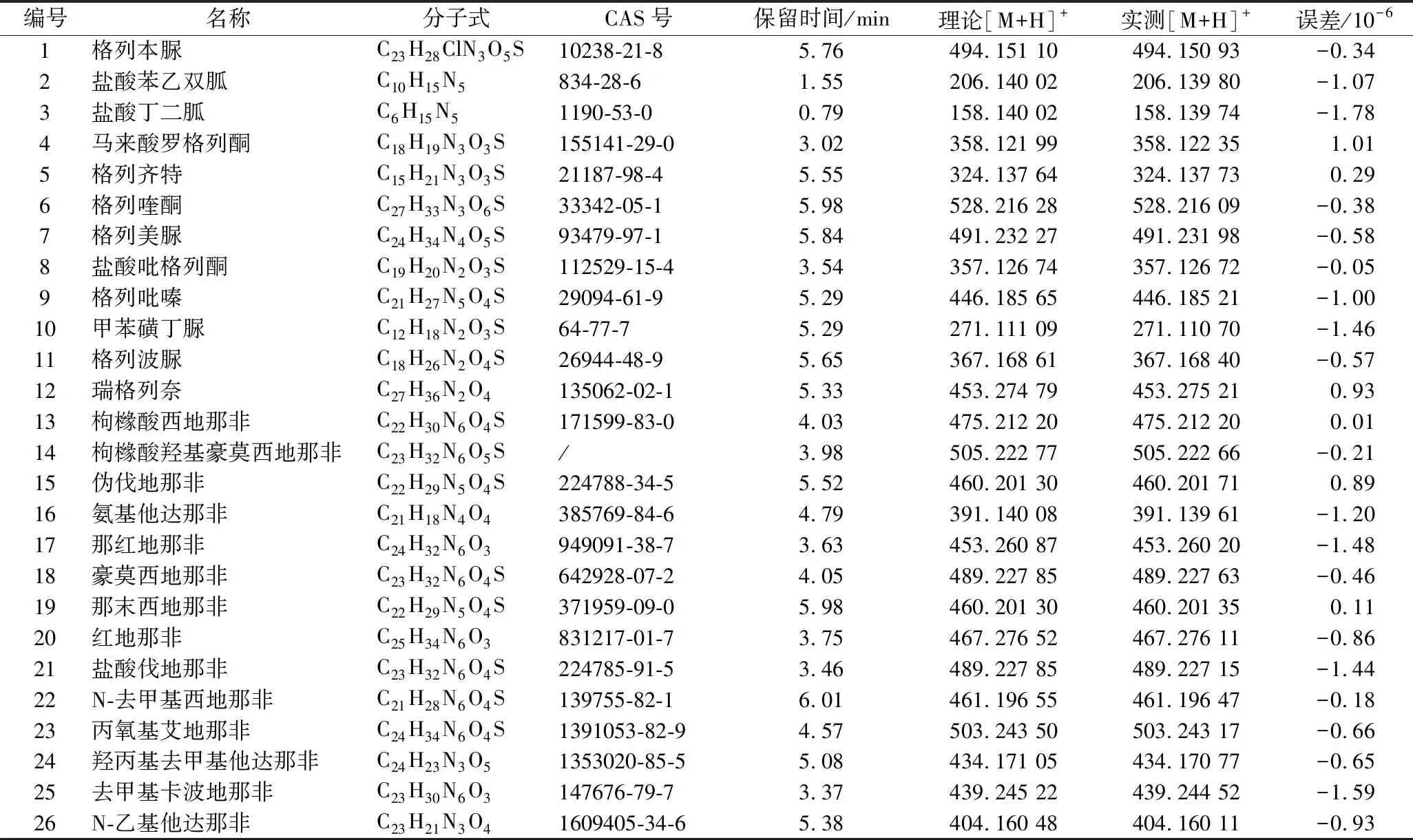
表1 26种非法添加化合物保留时间、理论质量数、实测质量数和质量数偏差

1-盐酸丁二胍;2-盐酸苯乙双胍;3-马来酸罗格列酮;4-去甲基卡波地那非;5-盐酸伐地那非;6-盐酸吡格列酮;7-那红地那非;8-红地那非;9-枸橼酸羟基豪莫西地那非;10-枸橼酸西地那非;11-豪莫西地那非;12-丙氧基艾地那非;13-氨基他达那非;14-羟丙基去甲基他达那非;15-格列吡嗪;16-甲苯磺丁脲;17-瑞格列奈;18-N-乙基他达那非;19-伪伐地那非;20-格列齐特;21-格列波脲;22-格列本脲;23-格列美脲;24-那末西地那非;25-格列喹酮;26-N-去甲基西地那非
通过以上条件的确认,建立了26种非法添加化合物的定性方法。Formula Predictor分子式预测软件可以实现基于一级精确质荷比、同位素丰度比的匹配定性,得分在70分以上,可以判定为疑似非法添加物。以格列喹酮为例,设定Formula Predictor软件的误差范围为5×10-6,元素组成为C、H、O、N、S以及元素数量分别为300、150、12、12、1,加和模式为[M+H]+。结果显示,C27H33N3O6S评分第一,匹配得分95.60,与格列喹酮的元素组成一致。图2为格列喹酮的XIC图。
图2 格列喹酮一级提取离子流图
按照“1.3.6数据库的建立”项下的条件,建立26种化合物的二级质谱图库。将样品的碎片离子信息与二级质谱图库中的碎片离子信息进行匹配,ACD Labs/2012软件根据匹配结果,给出相似度得分。通常,二级匹配得分大于60的非法添加物可判定为目标物。图3为格列喹酮的二级质谱匹配结果,样品主要的特征碎片与谱库中二级质谱图匹配良好,格列喹酮二级匹配得分为70.38。因此,可以确定样品中含有格列喹酮。根据欧盟SANTE/11945/2015指导文件[20],当使用高分辨质谱时,每个离子可以获得2个定性得分。本实验通过全扫描+数据依赖碎片扫描模式获得了一级的精确质荷比和丰富的二级碎片离子信息,格列喹酮二级质谱有16个定性得分,可实现精准确证。

a-样品二级质谱;b-二级质谱库格列喹酮
2.2 线性范围、标准曲线、检出限和定量限
本试验采用外标法绘制标准曲线,26种标准物质的线性方程、相关系数、检测限和定量限见表2。

表2 26种非法添加化合物的校准曲线、线性范围、相关系数、检出限及定量限
在各自线性范围内,26种目标化合物线性关系良好,相关系数r均达到0.99以上,检出限为0.000 1~0.010 9 mg/L,定量限为0.000 1~0.036 1 mg/L。
2.3 重复性实验
标准曲线浓度L6(0.072 5~0.313 7 mg/L)点按照1.3.3条件重复进样6次,结果显示,面积的RSD小于5%(表3),除了二甲双胍类降糖药保留较弱,出现了一些保留时间漂移,其余化合物保留时间的RSD均小于1.6%。

表3 26种非法添加化合物的保留时间及峰面积的相对标准偏差(n=6)
2.4 回收率及精密度实验
将不同质量浓度的标准液添加至阿卡波糖片中,制备不同添加浓度的样品,采用UHPLC-Q-TOF-MS法检测样品中26种目标物的回收率及RSD,每个浓度平行测定6次。结果显示,26种目标物的回收率为75.8%~128.4%之间,6份平行样品浓度的RSD均小于4%,具体结果见表4。
2.5 方法的实际应用
为证实本方法的实际应用价值,选用3份样品为研究对象,其中2份阴性对照和1份阳性对照作为本试验的质量控制样品。试验结果(图4)表明,阳性样品中m/z475.2122通道色谱峰保留时间为3.979 min,初步判断含有西地那非。二级质谱库检索,与西地那非相似度得分77.13,匹配度较高,说明该阳性对照检出西地那非。

表4 空白加标的回收率及相对标准偏差
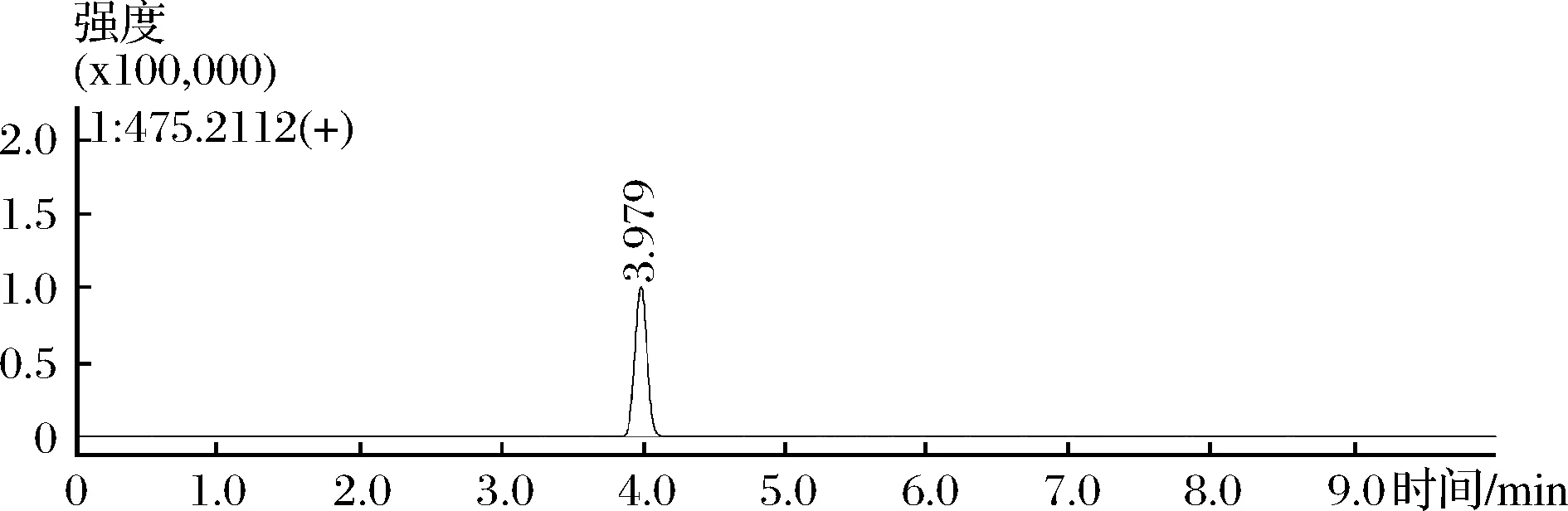
图4 m/z为475.212 2的样品提取离子流图
3 结论
本研究采用UHPLC-Q-TOF-MS技术,建立了26种降糖及抗疲劳药物的筛查方法。结果表明,26种降糖或抗疲劳类化合物分离度良好,通道无干扰。在各自线性范围内,相关系数r均达到0.99以上,检出限为0.000 1~0.010 9 mg/L,定量限为0.000 1~0.036 1 mg/L。回收率为75.8%~128.4%之间,6份平行样品浓度的RSD均小于4%,确认了目标化合物一级精确质荷比、保留时间、同位素丰度比等信息,建立了二级谱库,并应用于实际样品的分析。该方法回收率高、精密度良好、准确度高,可以满足降糖和抗疲劳类中成药和保健食品的非法添加物的筛查。本方法对于中成药和保健食品的质量安全控制具有重要意义。
猜你喜欢
保健与生活(2022年11期)2022-06-09
家庭医药(2022年2期)2022-02-18
中国土壤与肥料(2021年5期)2021-12-02
新体育(2020年1期)2020-01-13
吉林农业(2019年20期)2019-11-23
课程教育研究·新教师教学(2017年17期)2017-12-16
江苏农业科学(2017年6期)2017-05-11
中国实用医药(2017年3期)2017-03-20
