
Direct Detection of Target Genes in Viable Bacteria and Extracellular DNA Using Loop-Mediated Isothermal Amplification Assay
2020-03-25 06:02YANGQianqianZHANGXuzhiJIANGXiaoyuLIYangZHAOJunHAOZhihuiWANGPingpingQUKeming
渔业科学进展 2020年2期
关键词:小花
YANG Qianqian, ZHANG Xuzhi, JIANG Xiaoyu, LI Yang, ZHAO Jun, HAO Zhihui, WANG Pingping, QU Keming
Direct Detection of Target Genes in Viable Bacteria and Extracellular DNA Using Loop-Mediated Isothermal Amplification Assay
YANG Qianqian1,2, ZHANG Xuzhi2, JIANG Xiaoyu1,2, LI Yang2, ZHAO Jun2, HAO Zhihui3①, WANG Pingping3, QU Keming2①
(1. College of Marine Sciences, Shanghai Ocean University, Shanghai 201306; 2. Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Qingdao 266071; 3. School of Chemistry and Pharmaceutical Sciences, Qingdao Agriculture University, Qingdao 266109)
When the loop-mediated isothermal amplification (LAMP) assay is used for detecting target genes, DNA extraction is unnecessary in many cases. Simple pretreatment (e.g. heating) is enough to obtain rather sensitive responses. Even test samples without any pretreatment can be used as template. This feature suggests that LAMP is superior to PCR in developing point-of-care test strategies. In this study, usinggene fromas model, we verified that viable cells, dead cells and extracellular DNA could function as template in the LAMP assay. In the incubation at 63℃, viable bacteria in the LAMP reaction mixture lysed completely within 2 min, providing DNA template for nucleic acid amplification. Thegene in diluted culture medium, spiked tap water, spiked seawater and real seawater all could be detected, with or without the step of DNA extraction. We found that the complex substances in real sample (e.g. natural seawater) exhibited considerable inhibitory effect on the sensitivity of the LAMP assay. These outcomes are meaningful for building a point-of-care strategy by employing the LAMP assay for environmental monitoring, bio-resource surveys, food safety,. in particular those based on environmental DNA.
Loop-mediated isothermal amplification; DNA extraction-free; Direct gene detection; Viable cell; Extracellular DNA
1 Introduction
Nucleic acid amplification is increasingly used in a broad array of applications, such as molecular biology, clinical diagnostics, food safety, and environmental monitoring, to name a few. In particular, it is still a gold standard technique for analyzing samples with a small amount of nucleotides (Goda, 2015; Stedtfeld, 2014). Although polymerase chain reaction (PCR) is the first and remains the most popular amplification technology in various fields, it suffers from several drawbacks such as the requirement of multiple thermo- cycling steps, easy contamination, and high cost, which largely limit its application in resource-limited settings and, specially, in point-of-care use (Zhao, 2015).To address this issue, isothermal nucleic acid amplification techniques have recently been developed. In particular, loop-mediated isothermal amplification (LAMP), which has the potential to revolutionize molecular biology by reducing the need for highly sophisticated equipment, and by having low running costs and short turnaround times, is in bloom (Zhao, 2015; Zhang, 2014). LAMP shows higher specificity than PCR because it uses four to six different primers that bind to specific sites on the template strand. Moreover, the sensitivity of LAMP is less affected by substances that usually inhibit PCR reactions, such as food ingredients and blood components (Zhang, 2014; Kaneko, 2007; Abdul-Ghani, 2012; Kiddle, 2012; Wang, 2008). This suggests that simple assays could be developed using LAMP with the most cumbersome steps of sample pretreatment, such as DNA extraction and purification, eliminated (Safavieh, 2016; Njiru, 2012; Williams, 2017). Up to now, several studies have been reported on its capacity to directly amplify target genes from rapidly processed, crude sample matrix (Poon, 2006; Njiru, 2008; Priye, 2017; Bektas, 2016; Hayashida, 2015; Lee, 2016; Koizumi, 2012; Soejima, 2011; Ihira, 2010), and even original samples with or without simple mechanical-based pretreatment (Williams, 2017; Hill, 2008; Patterson, 2013; Enomoto, 2005; Kanitkar, 2017; Stedtfeld, 2016; Youn, 2016; Williams, 2017; Ahmad, 2017), considerably reducing the cost and turnaround time.
Target genes can be directly detected by employing LAMP assays without involving thesteps of DNA extraction and purification. For the success two key contributions should be acknowledged. First, the LAMP assay is tolerant of inhibition from complex substances in reaction matrices (Stedtfeld, 2014; Priye, 2017; Lee, 2016; Ahmad, 2017).Second, the acquired template plays a critical role. Some physical, chemical and biochemical methods, such as heat (Poon, 2006; Koizumi, 2012; Ihira, 2010), alkaline treatment (Bektas, 2016; Soejima, 2011) and addition of lysozyme (Lee, 2016), are sufficient for preparing DNA templates for LAMP. Furthermore, it has been reported that in some cases samples can be directly used as templates. This provides promise for simplifying the entire analysis operation. For example, Hill(2008)and Patterson(2013)found that unprocessed urine and whole blood could be added into LAMP reaction mixtures for the detection of target genes of specific bacteria. And in some other reports, the working mechanism has even been discussed preliminarily. Enomoto(2005)hypothesizedthat direct detection of HSV from swab samples originated from the large quantity of naked viral DNA as well as complete virions. Stedtfeldspeculated that adequate cell lysis occurred while incubating LAMP reactions at 63℃ (Kanitkar, 2017; Stedtfeld, 2016). They also found that heat treatment prior to incubation yields comparable levels of sensitivity to those of direct amplification without lysis. Youn(2016) postulated that target genes in dead and viable bacterial cells could both be amplified, and Williams(2017) postulated that extracellular DNA, larger cells and particulates all contributed to reactions when a suspension of crushed veliger was used as a template. However, to the best of our knowledge, until now there are no dedicated reports on the validation of working templates and the behavior of viable cells in the LAMP reaction.
Since the end of last century, novel strategies basedon the identification of DNA from environmental samples have proven noteworthy in detecting and monitoring not only common species, but also those that are endangered, invasive, or elusive (Williams, 2017; Ahmad, 2017; Bohmann, 2014; Giovannoni, 1990; Stoeck, 2010; Zielińska, 2017; Lee, 2017). In particular, the application of so-called environmental DNA (eDNA) analysis is shown to provide a potent tool for elucidating mechanistic insights in ecological and evolutionary processes (Bohmann, 2014). Though the identification of DNA is commonly done by PCR, now LAMP, which can speed up the implementation of management actions, either to protect or eradicate the organism of interest, is on the way (Lee, 2017). Generally, in test samples for gene analysis, there are not only viable cells, but also dead cells and extracellular DNA. Thus, to understand clearly what roles they play in the LAMP assay is of great significance for obtaining accurate target information. In this manuscript, we verified that viable cells, dead cells and extracellular DNA could each function as templates in LAMP assays using thegene fromas a model. In addition, we performed a series of experiments to determine why viable bacteria could work as templates during LAMP incubation. Inhibitory effects of the complex substances in natural seawater on the analysis sensitivity were also discussed.
2 Materials and methods
2.1 Culture and quantification of E. coli
The strain of(ATCC 43888) used in this study was purchased from BeNa Culture Collection Co. LTD (Beijing, China). Culture and quantification were performed according to the method we reported previously (Zhang, 2018). In brief, once the strain was taken from −80℃, it was pre-grown in liquid LB medium at 37℃ overnight with constant shaking. Next, cultures were inoculated into 200 ml of LB medium and incubated at 37℃ for 8~10 h to achieve mid-exponential phase. Then, the obtainedin the medium was diluted to desired concentrations immediately for further experiments. The densities (CFU/ml) of the un-dilutedcultures were calculated from the averages of colony counts and the magnitude of culture dilution by the plate counting method. Aqueous solutions were prepared with ultrapure water (Resistivity: 18.2 MΩ/cm) produced by a Poseidon-R70 water purification system (Research Scientific Instruments Co. LTD, Xiamen, China).
2.2 Preparation of DNA templates and LAMP primers
Genomic DNA templates were prepared according to the method we reported previously (Zhang, 2018; 2014). In brief, bacteria in the culture medium were collected by centrifugation. Then the genomic DNA was extracted using a Bacteria Genomic DNA Extraction Kit (Tiangen Biotech Co., LTD, Beijing, China) according to the manufacturer’s instructions.The purity was within the acceptable range of 1.8~2.0260 nm/280 nm. The stock concentration of the extracted genomic DNA (36.80 ng/μl) was determined with a Smartspec Plus spectrophotometer (Bio-Rad Lab, USA). Subsequent dilutions at the desired concentrationswere stored at 4℃ for no longer than one week prior to use.
Primers for detectinggene ofwere produced by Beijing SBS Genetech Co., LTD. (Beijing, China) with the following sequences (Table 1). The specificity of these primers has been confirmed previously by Zhao(2011)and Yan(2017).

Table 1 Primers used for detecting the Stx1gene of E. coli with the LAMP assay
2.3 Preparation of bacterial template for LAMP assays
Culturedwere filtered with Sterivex cartridges (SVGPL10RC,Millipore, Billerica, MA). Then, concentrated cells were released from the filter by adding 0.9 ml of elution buffer (Stedtfeld, 2014).Pure viable cells were obtained by the method of Youn(2016) with minor modifications.In brief, 400 μl of bacterial resuspension (108CFU/ml) and 100 μl of 0.5 mmol/L propidium monoazide (PMA, Biotium Inc., USA) were mixed. After 5 min incubation at room temperature in the dark, the mixtures were light-exposed for 15 min using a PMA-Lite™ LED photolysis device (Biotium Inc., USA) according to the manufacturer’s instructions. Before being used as template, the viable cells were carefully washed three times with PBS buffer to remove residual PMA, followed by resuspension with water and density calculation by plate counting. To obtain dead cells, the bacteria in the resuspension were heat-killed by exposure at 95℃ for 10 min (Ahmad, 2017). The death and viability of bacteria were confirmed by growth characterization.
2.4 LAMP assays
LAMP assays were performed as previous reported method with minor modifications (Zhang, 2014). In brief, a total volume of 25 μl reaction mixture containing 0.2 mmol/L each of F3 and B3, 0.8 mmol/L each of LoopF and LoopB, 1.6 mmol/L each of FIP and BIP, primers, 1.2 mmol/L each of deoxynucleotide triphosphate, 6 mmol/L MgSO4, 1 μl of 10×ThermoPol reaction buffer, 8 U ofDNA polymerase large fragment (NEB Co., LTD., USA), and 5 μl of template (viable cells, dead cells, extracted genomic DNA, aqueous samples without pretreatment,.), was incubated at 63℃ for 60 min in a thermal cycler (BioRad, Temecula, USA). Anelectrophoresis apparatus (DY-6, Xinghua Assay ApparatusFactory, Beijing, China) and a DNR bio-imaging systems(MF-ChemiBis 3.2, Israel) were used for electrophoresis analysis with 2.5% agarose gel. DL2000 DNA markers were purchased from TaKaRa Co., LTD (Dalian, China). Moreover, the fluorescent dye GeneFinder (Biov LTD., Xiamen, China) was used for visual characterization of LAMP reaction products. For each measurement, 3.68 pg extracted genomic DNA and ultrapure water were used as templates for positive and negative controls, respectively.
2.5 Spiked experiments
Unpurified tap water samples and natural seawater samples (collected from Jiaozhou Bay, China) were filtered with 0.22 μm Sterivex filters to remove bacteria followed by Silicone membranes (EMD Millipore Corp., Billerica, MA) to remove extracellular DNA (Stedtfeld, 2016). Then, bacteria or DNA were added to the filtered water to perform spiked experiments. Unless stated explicitly, all spiked water samples were used within 20 min to prevent the interference resulting cell lysis.
After incubation for ~10 h, a 1000-fold dilution of the initial culture medium was prepared with water, in which the density ofwas obtained by the plate counting method. The samples were prepared as follows: 1) To mock real samples, we added 100, 50, 20, 10, 5, 2 and 0 μl of a 1000-fold dilution (containing 109CFU/ml) into 19.900, 19.950, 19.980, 19.990, 19.995 and 19.998 ml filtered tap water or seawater. 2) To simulate extracellular DNA in test samples, extracted genomic DNA was added to filtered tap water or seawater at final concentrations of 2.2×100, 2.2×101, 2.2×102, 2.2×103, 2.2×104, 2.2×105and 0 fg/μl. 3) Bacteria suspensions (1012CFU/ml) of pure viable cells and dead cells were added into filtered tap water or seawater for final concentrations ranging from 0 to 107CFU/ml.
2.6 Detection of the Stx1 gene in natural seawater
Real seawater samples collected from Shilaoren Beach of Qingdao (120°28.308¢E; 36°5.502¢N) were transferred to the lab at ~8℃ and were used immediately. Thegenes in the samples were assayed by LAMP via three approaches: 1) Viable bacteria were collected as described above and used as template; 2) Five microliters of unpurified seawater sample were directly added to the reaction mixture; 3) After boiling treatment, 5 μl of the seawater sample was added into the reaction mixture. Note, in both of the second and third approach, 0.4×ThermoPol Reaction Buffer was used for LAMP to lower the final ionic strength.
2.7 Characterization of the survival behavior of viable cells during LAMP incubation at 63℃
An electrical bacterial growth sensor was used to characterize the survival behavior ofaccording to our previously published method (Zhang, 2018). First, to monitor bacterial growth, each disposable glass reaction tube was loaded with 1.3 ml sterile LB medium. Next, 20 μl of a suspension (containing 107CFU of pure viable cells) was added into the LAMP reaction mixture (200 μl in total), followed by incubation at 63℃. During the incubation, 10 μl of reaction mixture was transferred into a disposable glass reaction tube at 0, 1, 2, 3, 4 and 5 min, respectively. Disposable glass reactiontubes were simultaneously inserted into working channels of the electrical bacterial growth sensor. Apparent conductivity values were collected with the excitation frequency of 2.0 MHz, excitation amplitude of 16 V and recording rate of 10 second, and were used to generate growth curves by plotting against incubation time. Because the critical factor was to monitor conductivity changes during the process of bacterial growth rather than determination of the absolute value, an algorithm is presented here to obtain normalized apparent conductivity value (NACV), which was calculated as follows:
NACV=ACV−ACV0(1)
where NACVwas the real time normalized apparent conductivity value of bacterial culture medium in the tube obtained at point; ACVwas the apparent conductivity value collected at point; ACV0was the first apparent conductivity value collected at the beginning of the incubation. Namely, the apparent conductivity value at time = 0 was subtracted from all the following data during monitoring in order to normalize the starting growth levels at zero for all conditions. In addition, the survival behavior ofwas characterized by the plate culture method for comparison.
3 Results
3.1 LAMP assay of the Stx1 gene in mock samples
After an incubation of ~10 h, the density ofin the culture medium was 1.83×1012CFU/ml, as confirmed by the plate counting method. The following steps were used to prepare templates for the LAMP assays: 1) A 1000-fold dilution of the initial culture medium was prepared. 2) After the initial culture medium was passed through a 0.22 μm filter to remove bacteria and other large molecules, a 1000-fold dilution of the filtrate was prepared. 3) The filtrate (400 μl) was treated with PMA to make extracellular DNA non-amplifiable (Youn, 2016; Ahmad, 2017; Nocker, 2006).4) According to the method reported by Youn(2016), pure viable cells were obtained by eliminating amplifiable gene fragments from dead cells and extracellular DNA, and then they were re-suspended with water to prepare a bacterial suspension (107CFU/ml). 5) Half of the viable bacterial suspension was heated to prepare a dead cell suspension.
Five microliters of these five template preparations were added into respective reaction mixtures, and the LAMP assay was performed simultaneously in triplicate.The results were characterized by gel electrophoresis and the fluorescent dye GeneFinder, which has been proven to yield accurate quantification (Zhang, 2011). As shown in Fig.1, all of the amplification reactions were successful, with the exception of one with PMA-treated supernatant from diluted culture medium. Furthermore, the results of positive and negative control experiments indicated that the reaction system was reliable.
3.2 LAMP assay of the Stx1 gene in spiked samples
After a ~8 h incubation, a 1000-fold dilution of the initial culture medium, in which the density ofwas 9.95×108CFU/ml, was prepared. Then it was used to spike tap water samples as follows: 1) In the first group, nowas added. 2) In the second group, 50 μl of the 1000-fold dilution were added into 19.950 ml tap water. 3) Cells and other large molecules in 50 μl of 1000-fold dilution were removed by filtering. Then the filtrate was added to 19.950 ml tap water. 4) Pure viablein 50 μl of the 1000-fold dilution were obtainedfiltering and PMA-treatment, followed by addition to 19.950 ml tap water. 5) Pure viablein another 50 μl of 1000-fold dilution was killed by heating, also followed by addition to 19.950 ml tap water.
Five microliters of these five preparations were used as a template for each LAMP assay reaction. The gel electrophoresis and visualization results are shown in Fig.2. Thegene could clearly be detected in all spiked samples. Note, however, that there were not universal color changes in all three reaction tubes characterized with Genefinder, and that a ladder-like pattern of amplicons was present on one of the gel lanes. This phenomenon indicates that i) the sensitivity of the gel electrophoresis was higher than the sensitivity of the visual assay; and ii) there was a small amount of extracellular DNA in the unpurified tap water.

Fig.1 Detection of the Stx1 gene by LAMP assay with different templates
Results were determined by gel electrophoresis (a) and the fluorescent dye GeneFinder (b). In the positive control, 3.68 pg extracted genomic DNA was used as template, and in the negative control, pure water was used as template
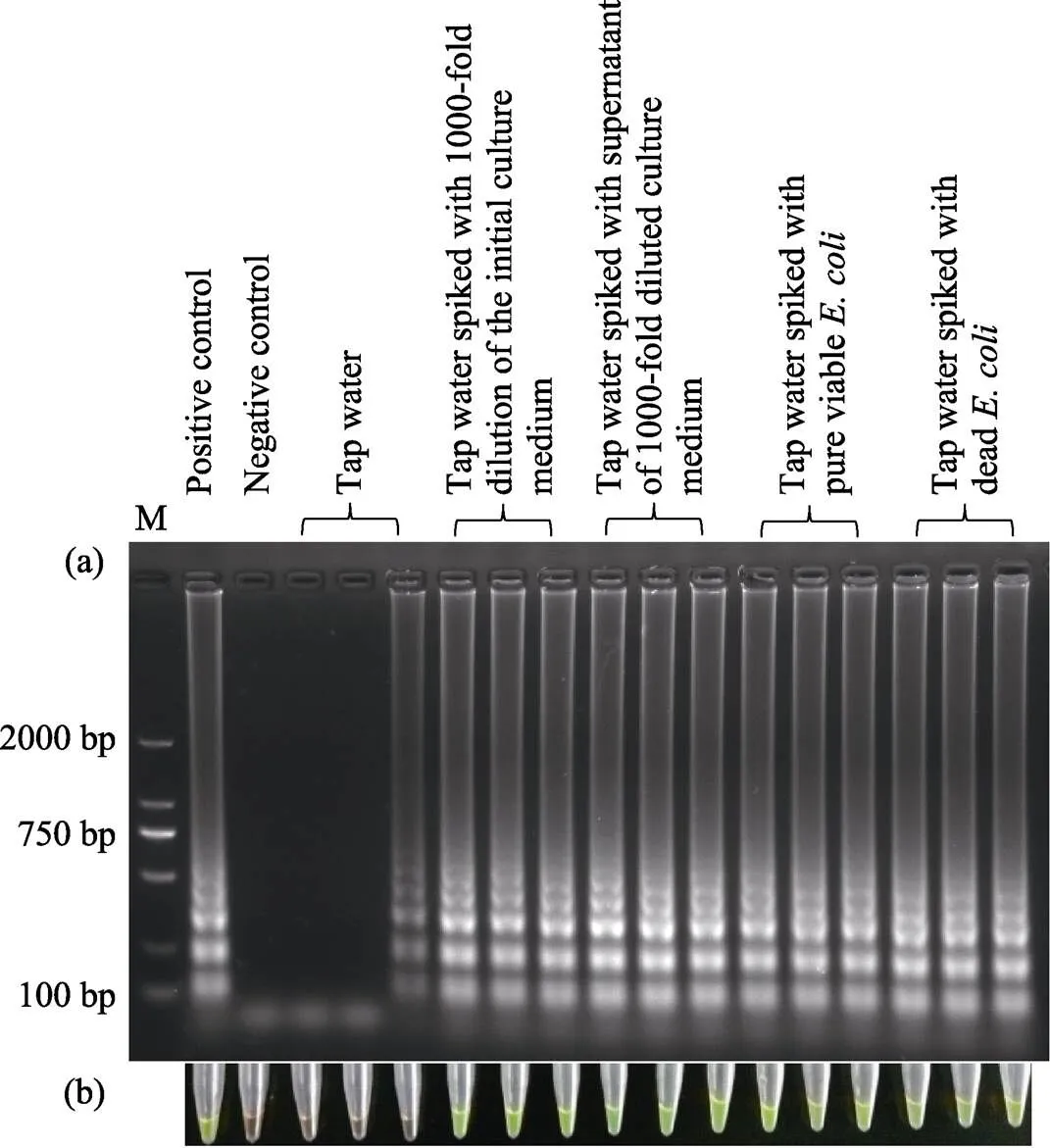
Fig.2 Detection of the Stx1 gene in spiked tap water samples by LAMP assay
Results were determined by gel electrophoresis (a) and the fluorescent dye GeneFinder (b). The positive and negative controls were the same in Fig.1. Pure water and 3.68 pg extracted genomic DNA were used as negative and positive template, respectively, in control experiments
Twenty milliliters of tap water samples were spiked with 1.99×104, 4.98×104, 9.95×104, 1.99×105, 4.98×105or 9.95×105CFUin culture medium. When 5 μl of the spiked samples were used as a template for the LAMP assay, thegene could be detected in all reactions, characterized both by gel electrophoresis and the fluorescent dye GeneFinder. Thegene could also be detected in the tap water samples, in which the final concentration of spiked genomic DNA ranged from 2.2×100to 2.2×105fg/μl. When tap water samples were spiked with pure viable bacteria at different concentrations, thegene was detectable (100%) in the case of ≥9.95×102CFU/ml, as characterized by gel electrophoresis.
After another incubation of ~8 h, a 1000-fold dilution of the initial culture medium was prepared in water, in which the density of thewas 1.07× 107CFU/ml. Then, it was used to spike seawater samples as follows: 1) In the first group, 50 μl of 1000- fold dilution were added to 19.950 ml clean seawater. 2) Cells and other large molecules in 50 μl of 1000-fold dilution were removed by filtering. Then the filtrate was added to 19.950 ml filtered seawater. 3) Pure viablein 50 μl of 1000-fold dilution were obtainedfiltering and PMA-treatment, followed by addition into 19.950 ml of filtered seawater. 4) The extracted genomic DNA was added into filtered seawater, allowing a final concentration of 2.2×105fg/μl.
Five microliters of these five solutions were used as template for the LAMP assay. The gel electrophoresis results are shown in Fig.3. Thegene could be detected in all spiked seawater samples. Twenty milliliters of filtered seawater samples were spiked with 2.14×104, 5.35×104, 1.07×105, 2.14×105, 5.35×105and 1.07×106CFUin culture media. When 5 μl of the spiked samples were used as template for the LAMP assay, thegene could be detected in all reactions, as characterized by gel electrophoresis. Thegene could also be detected in all of the spiked seawater samples in which the final concentration of genomic DNA was over the range of 2.2×102~2.2×105fg/μl. When seawater samples were spiked with pure viable bacteria at different concentration, thegene in 5 μl was detectable in all cases (100%) with ≥5.35× 103CFU/ml, as characterized by gel electrophoresis.
3.3 LAMP assay of the Stx1 gene in real seawater samples
The density of viablein the real seawater was calculated to be 19.20 CFU/ml after membrane filtration by the plate counting method. Pure viable bacteria in 100 ml of real seawater were obtainedfiltering and PMA-treatment, followed by resuspension in 100 μl PBS buffer. Then, 5 μl of the suspension was used as template for the LAMP assay to detect thegene. As shown in Fig.4 Lane 3~5, the ladder-like pattern of the amplicon was clear. Furthermore, when 5 μl of the natural seawater was directly used as template, with or without boiling-treatment, thegene could also be detected (Fig.4 Lane 6~11). It is worth noting that using undiluted seawater directly as template lowered the sensitivity of the LAMP assay remarkably, perhaps resulting from the inhibition caused by the salinity or complex matrices. We found that 0.4×ThermoPol Reaction Buffer worked well for the amplification task in this case. However, this parameter was not optimized further.
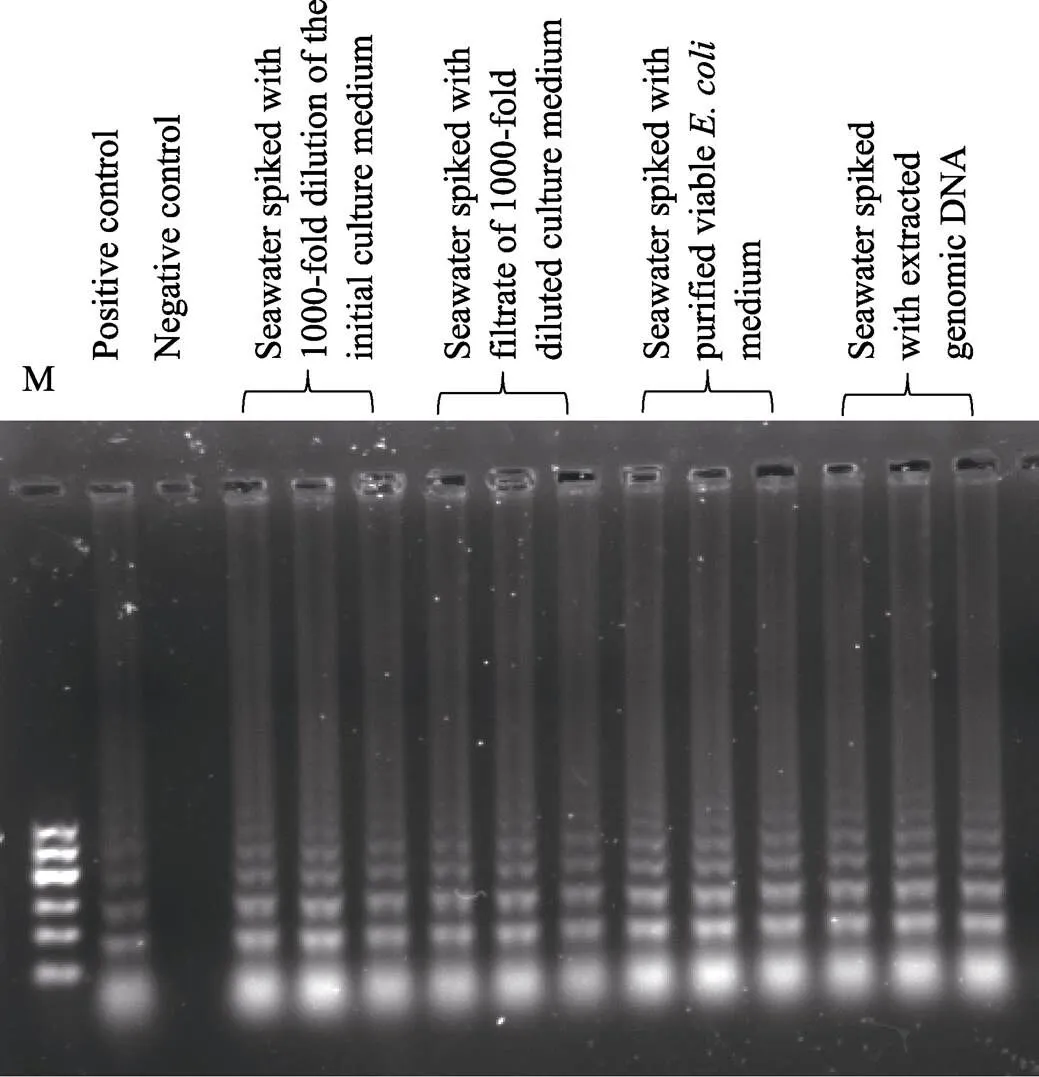
Fig.3 Detection of the Stx1 gene in spiked seawater samples by LAMP assay characterizing by gel electrophoresis
Pure water and 3.68 pg extracted genomic DNA were used as negative and positive template, respectively, in control experiments

Fig.4 Detection of the Stx1 gene in real seawater samples by LAMP assay using templates obtained with different pretreatment methods
Pure water and 3.68 pg extracted genomic DNA were used as negative and positive template, respectively, in control experiments
3.4 Survival behavior of viable cells during LAMP incubation at 63℃
Using the electrical bacterial growth sensor, we obtained typical curves ofgrowth (Fig.5a), which were generated by plotting the NACV against incubation time. When 10 μl of LAMP reaction mixture (containing 104CFU viable cells) without incubating at 63℃ was transferred into a disposable glass reaction tube for culture at 37℃ in the electrical bacterial growth sensor, there was an S-shaped growth curve. On the curve there was a lag phase, resulting from the stress that bacteria might experience after dilution and/or loading (Settu, 2015), as well as the time required for generating enough end products to produce detectable increasing conductivity. The lag phase was followed by an acceleration phase, during which the growth rate increased until a constant growth rate was achieved. Then, an exponential-like phase appeared, followed by a deceleration phase. These results reflect the typical growth of bacteria (Zhang, 2018), suggesting adequate vitality of the cells. The rest of the LAMP reaction mixture was incubated at 63℃. At the end of 1, 2, 3, 4 and 5 min, 10 μl of the mixture was transferred into disposable glass reaction tubes for culture at 37℃. A similar S-shaped growth curve was obtained when the inoculum was collected at the end of 1 min incubation, with a significantly increased time of detection (~600 min) compared to that of the inoculum that was collected before incubation at 63℃. This implies that there were still viable cells in the culture medium; nevertheless, the number was much less than that sampled from the LAMP mixture before incubation (Zhang, 2018).If 10 μl of inoculum was sampled from the LMAP mixture with incubation at 63℃ for more than 1 min, a horizontal line was obtained due to unchanged conductivity, indicating a nonoccurrence of bacterial growth. In other words, the cells all died completely. The survival behavior ofwas also characterized by the plate counting method (Fig.5b). The results were in good agreement with those obtained with the electrical bacterial growth sensor.
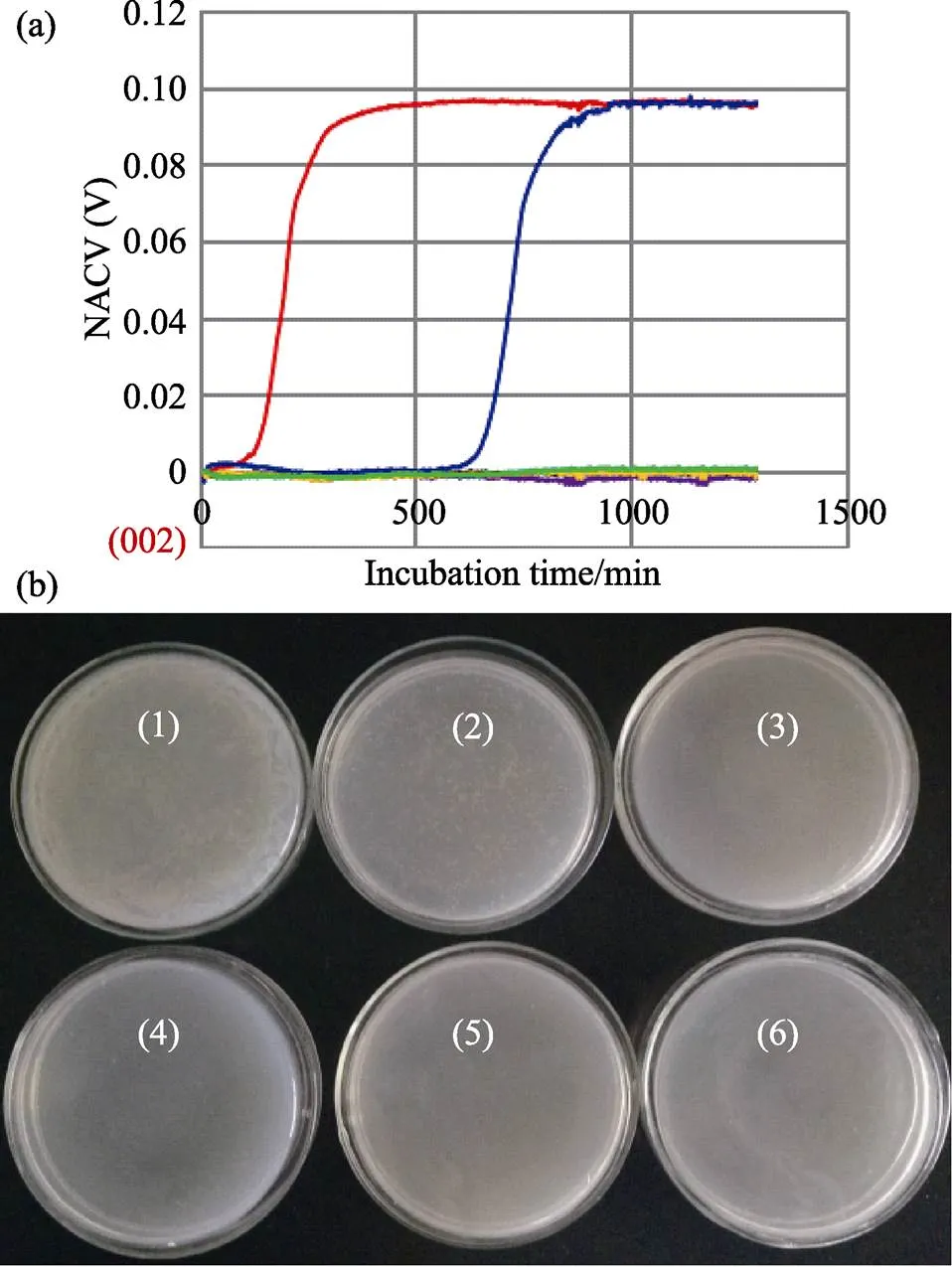
Fig.5 Growth characterization for studying survival behavior of viable cells
(a) Growth curves of(NACV. incubation time) in LB medium obtained with the electrical bacterial growth sensor. For the S-shaped curve in red, the inoculum was 10 μl of LAMP reaction mixture (containing 104CFU viable cells), which was sampled before incubation at 63℃. For the S-shaped curve in blue, the inoculum was 10 μl of the LAMP reaction mixture, which was sampled after 1 min incubation at 63℃. For the other horizontal lines, the inoculum was 10 μl of the LAMP reaction mixture sampled at the end of 2, 3, 4 and 5 min incubation. In each disposable glass reaction tube 1.3 ml LB medium was loaded. The operation parameters were as follows: excitation frequency of 2.0 MHz; excitation amplitude of 16 V; recording rate per 10 s. (b) Photographs ofcolonies. For plates number 1 to 6 the inoculum was 10 μl of LAMP reaction mixture containing viable cells, which was sampled at the end of 0, 1, 2, 3, 4 and 5 min of incubation at 63℃, respectively
4 Discussion
The detection of target genes with PCR techniques commonly requires cell lysis, DNA extraction and template purification; necessitates the use of bulky instrumentations; and remains cumbersome (Stedtfeld, 2014; Enomoto, 2005). LAMP has the potential to circumvent these issues because of the reduced dependency on pretreatment of the test samples (Safavieh, 2016; Njiru, 2012; Williams, 2017), lowering the need for highly sophisticated equipment and turnaround times (Zhao, 2015; Zhang, 2014). However, to avoid false negatives and overestimation of viable cell numbers when performing LAMP, greater understanding of the role of templates is still needed at present.
We found that a 1000-fold diluted filtrate of culturemedium in which cells had been removed(Kanitkar, 2017; Stedtfeld, 2016),could be used as template for the LAMP assay to detect thegene, indicating that in this case 1) there was extracellular DNA ofin the filtrate, as a result of natural lysis of dead cells or active secretion of viable cells (Biller, 2017); and 2) the reaction system was tolerant to the substances introduced by the culture medium. Upon exposure to blue light, PMA can intercalate into double stranded DNA and form covalent linkages, resulting in chemically modified DNA, which cannot be amplified by PCR or LAMP reactions (Youn, 2016; Nocker, 2006; Chen, 2011). Therefore, when this template, i.e. the 1000-fold diluted filtrate of culture medium, was treated with PMA, thegene could not be detected by the LAMP assay. This suggested that in this case all of the extracellular DNA were non-amplifiable. We considered the adequate PMA making the difference from the phenomenon found by Ahmad(2017).
Our results confirmed that in culture medium at exponential phase there were both viable cells and extracellular DNA. Pure viable cells, dead cells, extracellular DNA and their mixture in culture medium could each be used as templates for the LAMP assay, reconfirming the non-ignorable effects of coexisting extracellular DNA in determining viable cells by DNA-based molecular detection techniques (Ahmad, 2017; Chen, 2011).Even when all of the cells were removed by filtration, thegene in tap water could be sensed by the LAMP assay as determined by gel electrophoresis. Using DNA extraction-free LAMP for detecting thegene in groundwater samples, Stedtfeld(2014) found that filtration of larger volumes caused loss of sensitivity, which they attributed to the loss of extracellular DNA that could pass through the cartridge. Our results confirmed this hypothesis.
The results of spiked experiments also showed that the target gene in pure viable cells, dead cells and extracellular DNA could be detected by the LAMP assay, whether it was from tap water or seawater. While the detectable concentrations of spiked genomic DNA in tap water and seawater were over the range of 2.2×100~2.2×105fg/μl and 2.2×102~2.2×105fg/μl, respectively, the detection limits of pure viable bacteria in tap water and seawater were 9.95×102CFU/ml and 5.35×103CFU/ml, respectively, suggesting that substances in seawater showed considerable inhibitory effects on LAMP reactions, as they did in the culture medium (Enomoto, 2005) and other real and mock samples (Stedtfeld, 2014; Feng, 2018).Using 5 μl of seawater as template in which the concentration of spiked pure viable bacteria was 5.35×100CFU/ml, we did not have successful amplification. However, when the cells from 100 ml of this spiked seawater were collected by filtering, followed by resuspension, the target gene could be detected clearly, suggesting that a step of enrichment by filtering (Stedtfeld, 2014; Stedtfeld, 2016) or centrifugation (Kanitkar, 2017) was an effective approach for detecting target genes at low content samples, while omission of DNA extraction could save both time and labor in preparing templates for the LAMP assay.
Among the reports of direct LAMP, performed without DNA extraction and purification steps, heat-treatment of samples was considered to benefit the reactions due to the increased cell lysis (Poon, 2006; Njiru, 2008; Ihira, 2010). However, Stedtfeld(2016) as well as Ahmad(2017)found that heat-treatment of samples prior to LAMP had no observable influence on the time at which the SNR reached an arbitrary cut-off of 10 (t) or sensitivity compared to direct amplification without lysis. In particular, viable cells could be used as template directly. There had been various hypotheses for how they work. Ahmad(2017) considered that the success of the LAMP assay might be due to increased cell permeability at a temperature of 63℃, providing access to the genetic contents in the reaction solution. Stedtfeld(2014) thought that perhaps the smallerpolymerase had a high rate of passage into intact bacterial cells, or maybe cells become porous at the amplification reaction temperature of 63℃. Kanitkar(2017) hypothesized that cell lysis occurred while incubating LAMP reactions at 63℃, though they didn´t think the lysis was adequate. Until now, the mechanisms of unintentional lysis were unclear. The results of our survival experiments showed that the number of viable cells in the LAMP reaction mixture only slightly decreased after incubation at room temperature for 10 min but decreased sharply during incubation at 63℃. All of the viable cells in the LAMP mixture died completely when the incubation was more than 2 min. However, in control experiments, in which water was used in place of the LAMP mixture, more than half of the spiked viablesurvived at the end of 10 min. These results confirmed that viable cells were killed rapidly in the LAMP reaction mixture during the incubation at 63℃. This discovery explained the moderate influence ontand the sensitivity of heat-treatment prior to LAMP (Stedtfeld, 2016; Ahmad, 2017). But it was unclear now which species were at work for the killing.
Shipping of samples to an off-site laboratory is cumbersome, costly, and requires proper disposal, which influences the number of replicates that are generally analyzed. Transportation of samples also causes chances for leaking/contamination, and increased storage time has the potential to alter the integrity of unstable biomarkers (Stedtfeld. 2014). Thus, the validation of the LAMP assay, which can be performed at the point of care due to the ability to use test samples directly as template, has potential benefits for multiple researchers. In particular, the application of eDNA analysis for ecological research, such as the distribution of aquatic and terrestrial organisms and their biodiversity, is becoming increasingly more popular (Williams, 2017; Bohmann, 2014; Kristy, 2016; Shan, 2018). The combination of eDNA with direct LAMP may provide a beneficial combination.From our results using spiked experiments and the detection of thegene in real seawater samples, we could see that the omission of DNA extraction and purification saved both time and labor (Enomoto, 2005). However, this was achieved at the expense of sensitivity and accuracy, due to variation in the extracellular DNA, as well as inhibitory factors. Furthermore, quantitative correlation between the copy number of marker genes in eDNA and the number of target organisms commonly is unstable (Williams, 2017). Therefore, to design a strategy for field-based analysis, these factors are worthy of additional consideration.
We confirmed that viable cells, dead cells and extracellular DNA all can function as template for the LAMP assay, using thegene ofas a model. Thus, to detect similar target bacteria in samples, extracellular DNA could potentially be captured with silica membranes (Harikai, 2015)as an alternative to collecting cells by filtration (Stedtfeld, 2014).Moreover, in the case of quantifying viable cells, before determining target genes with LAMP, analysts should take additional steps, such as PMA-treatment, to eliminateinterference from co-existing extracellular DNA. Otherwise the quantities might be overestimated.
Samples (diluted culture medium, tap water and seawater) can be directly used as template for the LAMP assay without physical, chemical or biochemical pretreatment, due to the presence of viable cells, dead cells or extracellular DNA. Direct amplification from unpurified samples could reduce the complexity of the gene analysis instruments, as well as difficulties of operation, allowing for simple, in situ, rapid and cost- effective gene analysis. However, common pretreatment approaches, e.g., concentration and purification, are useful for higher sensitivity and accuracy. This should be considered carefully when designing experiments for detecting target bacteria in specific samples, especially at low concentration. In addition, when researchers combine LAMP and eDNA, the quantitative correlation between the copy number of marker genes in eDNA and the target number of organisms should be closely considered.
5 Conclusion
Using thegene fromas model, we verified that viable cells, dead cells and extracellular DNA could function as template in the LAMP assay. The target gene in viablecan be amplified due to the efficient lysis of cells in the LAMP reaction mixture during the incubation at 63℃, allowing for replication of the target nucleic acid in the presence ofDNA polymerase. Therefore, heat-treatment is not necessary prior to LAMP assay. In addition, thegene in diluted culture medium, spiked tap water, spiked seawater and real seawater all could be detected, with or without the step of DNA extraction. However, the complex substances in real sample (e.g. natural seawater) exhibited considerable inhibitory effect on the sensitivity of the LAMP assay. These outcomes are meaningful for building a point-of-care strategy by employing the LAMP assay for environmental monitoring, bio-resource surveys, food safety, etc. in particular those based on environmental DNA.
Conflict of interest
The authors declare that they have no conflict of interest.
Abdul-Ghani R, Al-Mekhlafi AM, Karanis P. Loop-mediated isothermal amplification (LAMP) for malarial parasites of humans: Would it come to clinical reality as a point-of-care test? Acta Tropica, 2012, 122(3): 233–240
Ahmad F, Stedtfeld RD, Waseem H,. Most probable number-loop mediated isothermal amplification (MPN-LAMP) for quantifying waterborne pathogens in <25 min. Journal of Microbiological Methods, 2017, 132: 27–33
Bektas A, Chapela I. Efficiency of a fluorescent, non-extraction LAMP DNA amplification method: Toward a field-based specific detection of maize pollen grains. Aerobiologia, 2016, 32(3): 481–488
Biller SJ, Mcdaniel LD, Breitbart M,. Membrane vesicles in sea water: Heterogeneous DNA content and implications for viral abundance estimates. ISME Journal, 2017, 11(2): 394–404
Bohmann K, Evans A, Gilbert MT,. Environmental DNA for wildlife biology and biodiversity monitoring. Trends in Ecology & Evolution, 2014, 29(6): 358–367
Chen S, Wang F, Beaulieu JC,. Rapid detection of viable salmonellae in produce by coupling propidium monoazide with loop-mediated isothermal amplification. Applied and Environmental Microbiology, 2011, 77(12): 4008–4016
Enomoto Y, Yoshikawa T, Ihira M,. Rapid diagnosis of herpes simplex virus infection by a loop-mediated isothermal amplification method. Journal of Clinical Microbiology, 2005, 43(2): 951–955
Feng N, Zhou Y, Fan Y,. Yersinia pestisdetection by loop-mediated isothermal amplification combined with magnetic bead capture of DNA. Brazilian Journal of Microbiology, 2018, 49(1): 128–137
Giovannoni SJ, Britschgi TB, Moyer CL,. Genetic diversity in Sargasso Sea bacterioplankton. Nature, 1990, 345: 60–63
Goda T, Tabata M, Miyahara Y. Electrical and electrochemical monitoring of nucleic acid amplification. Frontiers in Bioengineering and Biotechnology, 2015, 3: 29
Harikai N, Shinomiya K. Application of an alkaline and silica membrane DNA extraction method to detect mitochondrial DNA in foods. Food Analytical Methods, 2015, 8(5): 1215– 1224
Hayashida K, Kajino K, Hachaambwa L,. Direct blood dry LAMP: A rapid, stable, and easy diagnostic tool for human African trypanosomiasis. PLoS Neglected Tropical Diseases 2015, 9(3): e0003578
Hill J, Beriwal S, Chandra I,. Loop-mediated isothermal amplification assay for rapid detection of common strains of. Journal of Clinical Microbiology, 2008, 46(8): 2800
Ihira M, Sugiyama H, Enomoto Y,. Direct detection of human herpesvirus 6 DNA in serum by variant specific loop-mediated isothermal amplification in hematopoietic stem cell transplant recipients. Journal of Virological Methods, 2010, 167(1): 103–106
Kaneko H, Kawana T, Fukushima E,. Tolerace of loop-mediated isothermal amplification to a culture medium and biological substances. Journal of Biochemical and Biophysical Methods, 2007, 70(3): 499–501
Kanitkar YH, Stedtfeld RD, Hatzinger PB,. Development and application of a rapid, user-friendly, and inexpensive method to detectsp. reductive dehalogenase genes from groundwater. Applied Microbiology and Biotechnology, 2017, 101: 4827–4835
Kiddle G, Hardinge P, Buttigieg N,. GMO detection using a bioluminescent real time reporter (BART) of loop mediated isothermal amplification (LAMP) suitable for field use. BMC Biotechnology, 2012, 12(1): 15
Koizumi N, Nakajima C, Harunari T,. A new loop-mediated isothermal amplification method for rapid, simple, and sensitive detection ofspp. in urine. Journal of Clinical Microbiology, 2012, 50(6): 2072–2074
Kristy D, Fronhofer EA, Elvira M,. Environmental DNA reveals that rivers are conveyer belts of biodiversity information. Nature Communications, 2016, 7: 12544
Lee D, Kim YT, Lee JW,. An integrated direct loop-mediated isothermal amplification microdevice incorporated with an immunochromatographic strip for bacteria detection in human whole blood and milk without a sample preparation step. Biosensors and Bioelectronics, 2016, 79: 273–279
Lee PL. DNA amplification in the field: Move over PCR, here comes LAMP. Molecular Ecology Resources, 2017, 17(2): 138–141
Njiru ZK,. Loop-mediated isothermal amplification (LAMP)method for rapid detection of Trypanosoma brucei rhodesiense. PLoS Neglected Tropical Diseases, 2008, 2: e147
Njiru ZK. Loop-mediated isothermal amplification technology: Towards point of care diagnostics. PLoS Neglected Tropical Diseases, 2012, 6(6): e1572
Nocker A, Cheung CY, Camper AK. Comparison of propidium monoazide with ethidium monoazide for differentiation of live vs. dead bacteria by selective removal of DNA from dead cells. Journal of Microbiological Methods, 2006, 67(2): 310–320
Patterson AS, Heithoff DM, Ferguson BS,. Microfluidic chip-based detection and intraspecies strain discrimination of salmonella serovars derived from whole blood of septic mice. Applied and Environmental Microbiology, 2013, 79(7): 2302–2311
Poon LL, Wong BW, Ma EH,. Sensitive and inexpensive molecular test for falciparum malaria: detecting Plasmodium falciparum DNA directly from heat-treated blood by loop-mediated isothermal amplification. Clinical Chemistry, 2006, 52(2): 303–306
Priye A, Bird SW, Light YK,. A smartphone-based diagnostic platform for rapid detection of Zika, chikungunya, and dengue viruses. Scientific Reports, 2017, 7: 44778
Safavieh M, Kanakasabapathy MK, Tarlan F,. Emerging loop-mediated isothermal amplification-based microchip and microdevice technologies for nucleic acid detection. ACS Biomaterials Science & Engineering, 2016, 2(3): 278
Settu K, Chen CJ, Liu JT,. Impedimetric method for measuring ultra-lowconcentrations in human urine. Biosensors and Bioelectronics, 2015, 66: 244–250
Shan X, Li M, Wang W. Application of environmental DNA technology in aquatic ecosystem. Progress in Fishery Sciences, 2018, 39(3): 23–29
Soejima M, Egashira K, Kawano H,. Rapid detection of haptoglobin gene deletion in alkaline-denatured blood by loop-mediated isothermal amplification reaction. Journal of Molecular Diagnostics, 2011, 13(3): 334–339
Stedtfeld R, Stedtfeld TM, Kronlein M,. DNA extraction- free quantification of Dehalococcoides spp. in groundwater using a hand-held device. Environmental Science & Technology, 2014, 48(23): 13855
Stedtfeld RD, Stedtfeld TM, Samhan F,. Direct loop mediated isothermal amplification on filters for quantification of Dehalobacter in groundwater. Journal of Microbiological Methods, 2016, 131: 61–67
Stoeck T, Bass D, Nebel M,. Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water. Molecular Ecology, 2010, 19: 21–31
Wang L, Shi L, Alam MJ,. Specific and rapid detection of foodborne Salmonella by loop-mediated isothermal amplification method. Food Research International, 2008, 41(1): 69–74
Williams MR, Stedtfeld RD, Engle C,. Isothermal amplification of environmental DNA (eDNA) for direct field-based monitoring and laboratory confirmation ofsp. PLoS One, 2017, 12(10): e0186462
Williams MR, Stedtfeld RD, Waseem H,. Implications of direct amplification for measuring antimicrobial resistance using point-of-care devices. Analytical Methods, 2017, 9(8): 1229–1241
Yan M, Li W, Zhou Z,. Direct detection of various pathogens by loop-mediated isothermal amplification assays on bacterial culture and bacterial colony. Microbial Pathogenesis, 2017, 102: 1–7
Youn SY, Jeong OM, Choi BK,. Application of loop-mediated isothermal amplification with propidium monoazide treatment to detect live Salmonella in chicken carcasses. Poultry Science, 2016, 96: 458–464
Zhao X, Li Y, Wang L,. Development and application of a loop-mediated isothermal amplification method on rapid detectionO157 strains from food samples. Food & Fermentation Industries, 2011, 37(5): 2183–2188
Zhao Y, Chen F, Li Q,. Isothermal amplification of nucleic acids. Chemical Reviews, 2015, 115(22): 12491
Zhang X, Liu W, Lu X,. Monitoring the progression of loop-mediated isothermal amplification using conductivity. Analytical Biochemistry, 2014, 466: 16–18
Zhang X, Lowe SB, Gooding JJ. Brief review of monitoring methods for loop-mediated isothermal amplification (LAMP). Biosensors and Bioelectronics, 2014, 61(20): 491–499
Zhang X, Qu K, Li Q,. Recording the reaction process of loop-mediated isothermal amplification (LAMP) by monitoring the voltammetric response of 2´-deoxyguanosine 5´-triphosphate. Electroanalysis, 2011, 23(10): 2438–2445
Zhang X, Jiang X, Yang Q,. Online monitoring of bacterial growth with electrical sensor. Analytical Chemistry, 2018, 90(10): 6006–6011
Zielińska MS, Kidawa DD, Stempniewicz PL,. Environmental DNA as a valuable and unique source of information about ecological networks in Arctic terrestrial ecosystems. Environmental Reviews, 2017, 25: 282–291
2019-02-20,
2019-03-05
2095-9869(2020)02-0041-10
Chinese Library Classification No. S965.3
A
10.19663/j.issn2095-9869.20190220002
http://www.yykxjz.cn/
Yang QQ, Zhang XZ, Jiang XY, Li Y, Zhao J, Hao ZH, Wang PP, Qu KM. Direct detection of target genes in viable bacteria and extracellular DNA using loop-mediated isothermal amplification assay. Progress in Fishery Sciences, 2020, 41(2): 41–50
* This work was supported by the Special Scientific Research Funds for Central Non-Profit Institutes, Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences (20603022018020; 20603022016003). YANG Qianqian, E-mail: 2238854767@qq.com
Prof. QU Keming, E-mail: qukm@ysfri.ac.cn; HAO Zhihui, E-mail: abplab@126.com
(编辑 冯小花)
猜你喜欢
小学生学习指导(当代教科研)(2021年6期)2021-05-23
黄河之声(2021年2期)2021-03-29
学生天地(2020年11期)2020-08-25
作文周刊·小学二年级版(2019年32期)2019-10-15
阅读(低年级)(2019年8期)2019-10-10
歌海(2019年2期)2019-06-11
小学生学习指导(低年级)(2018年3期)2018-01-31
创新作文(1-2年级)(2017年5期)2017-12-07
红蜻蜓(2016年6期)2016-05-14
