
氟丙嘧草酯原药高效液相色谱分析
2020-03-19 06:57张继伟牛洺鑫王鹏思
世界农药 2020年2期
张继伟,牛洺鑫,王鹏思,张 博
(中化化工科学技术研究总院有限公司,北京 100083)
氟丙嘧草酯(butafenacil,CAS登录号134605-64-4)是先正达公司开发的非选择性脲嘧啶类除草剂,商品名称Inspire,其混剂Rebin GT为氟丙嘧草酯+草甘膦。试验表明,氟丙嘧草酯作为非选择性除草剂,对于某些杂草有优异的防效[1]。虽然目前氟丙嘧草酯原药和相关制剂还没有在国内登记,但是对原药的工艺研究和生物活性研究一直在进行[2-7],由此可见,该产品在国内的关注度较高,未来有望得以登记并商业化。本文采用高效液相色谱法对氟丙嘧草酯原药的定性、定量分析进行了研究,首次公开报道了一种快速、简便、准确的氟丙嘧草酯原药分析方法,为后期该产品及其相关制剂配方的分析研究提供理论依据。
1 试验部分
1.1 材料与试剂
99.0 %氟丙嘧草酯标样(德国 Dr.Ehrenstorfer公司);水:纯净水(屈臣氏);乙腈:色谱纯(Thermo Fisher);醋酸:分析纯(国药集团);氟丙嘧草酯原药样品(北京颖泰嘉和生物科技有限公司)。
1.2 仪器与设备
高效液相色谱仪:安捷伦1200高效液相色谱仪,配有紫外检测器和自动进样器;色谱柱:Zorbax Eclipse XDB-C18色谱柱(150×4.6 mm,5µm);超声波清洗器(昆山市超声仪器有限公司);0.45 μm有机相针式过滤器(津腾);50 mL容量瓶(北京化玻站有限公司)。
1.3 液相色谱操作条件
流动相:乙腈-0.5%醋酸水溶液(体积比60.0∶40.0);流速:1.0 mL/min;检测波长:272 nm;柱温:30 ℃;进样体积:5.0 μL。保留时间:约6.2 min。氟丙嘧草酯原药的高效液相色谱图见图1。
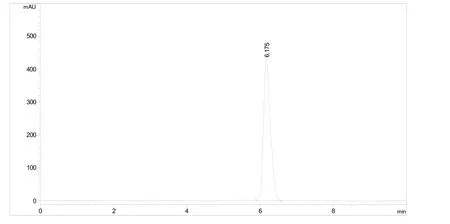
图1 氟丙嘧草酯原药的高效液相色谱图
1.4 测定步骤
1.4.1 标样溶液的配制
称取0.05 g氟丙嘧草酯标样(精确至0.000 2 g),置于50 mL容量瓶中,加入适量乙腈,在超声波清洗器上超声10 min,使标样全部溶解,冷却至室温后,用乙腈准确定容至标线,摇匀,用0.45 μm滤膜过滤,备用。按照上述方法分别配制标样溶液1和标样溶液2。
1.4.2 试样溶液的配制
称取0.05 g(精确至0.000 2 g)氟丙嘧草酯原药试样,置于50 mL容量瓶中,加入适量乙腈,在超声波清洗器上超声10 min,使试样全部溶解,冷却至室温后,用乙腈准确定容至标线,摇匀,用0.45 μm滤膜过滤,备用。按照上述方法分别配制试样溶液1和试样溶液2。
1.5 测定
在上述操作条件下,待仪器基线稳定后,连续进数针标样溶液,直至相邻2针氟丙嘧草酯标样的响应值相对变化小于1.5%后,按照标样溶液1、试样溶液1、试样溶液1、标样溶液1、标样溶液2、试样溶液2、试样溶液2、标样溶液2的顺序进行测定。
1.6 计算
将测得的2针试样溶液中氟丙嘧草酯峰面积以及试样前后2针标样溶液中氟丙嘧草酯响应因子分别进行平均。试样中氟丙嘧草酯的质量分数ω1(%)按式(1)和(2)计算:
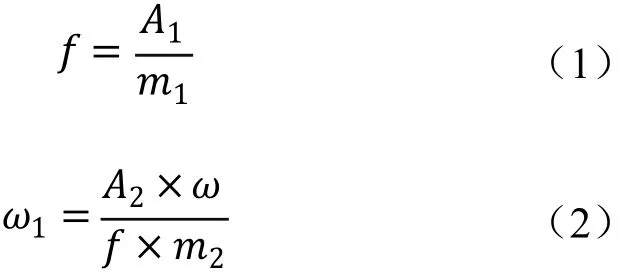
式中:f为标样溶液中氟丙嘧草酯的响应因子;m1为氟丙嘧草酯标样的质量,g;A1为标样溶液中氟丙嘧草酯峰面积的平均值;ω1为试样中氟丙嘧草酯的质量分数,%;A2为试样溶液中氟丙嘧草酯峰面积的平均值;ω为氟丙嘧草酯标样的纯度,%;m2为试样的质量,g。
将试样溶液1和试样溶液2中测得的氟丙嘧草酯质量分数的平均值作为测定结果。
2 结果与讨论
2.1 检测波长的选择
在190~380 nm的波长范围内,对氟丙嘧草酯进行紫外扫描,得到其相应的吸收波长与响应值的紫外吸收光谱图。发现氟丙嘧草酯在 272 nm波长有最大紫外吸收,杂质干扰小,且流动相无吸收。因此,综合考虑多种因素后,将检测波长选定为272 nm。
2.2 流动相的选择
采用安捷伦Zorbax Eclipse XDB-C18(150×4.6 mm,5µm)色谱柱进行反相液相色谱分析。依据氟丙嘧草酯自身的理化性质,采用溶解性能较好的乙腈-水体系作为流动相。试验结果表明当仅使用乙腈-水体系时,检测峰形有拖尾现象,故在水相中加入了千分之五的醋酸来调节流动相pH,以便改善出峰拖尾现象。通过对不同配比的乙腈-0.5%的醋酸水体系的分离效果对比,确定最佳的流动相配比为乙腈-0.5%醋酸水体积比60.0∶40.0,流速为1.0 mL/min。此时,有效成分的保留时间为6.2 min左右,检测峰形尖锐且对称,没有拖尾现象。此方法达到了快速简便分离的要求,是一种理想的氟丙嘧草酯原药分析方法。
2.3 方法线性相关性的测定
以乙腈为溶剂,分别配制质量浓度约为0.400g/L、0.700 g/L、1.000 g/L、1.100 g/L、1.300 g/L、1.600 g/L的氟丙嘧草酯标样溶液。按照上述色谱条件,依次进行测定。将测得的结果,以进样质量浓度为横坐标,不同质量浓度溶液的峰面积为纵坐标,绘制氟丙嘧草酯的线性相关曲线。结果表明:氟丙嘧草酯在0.400~1.600 g/L呈良好的线性关系,其线性方程为y=5 585.1x+35.178,线性相关系数R2为0.999 8。
2.4 方法精密度的测定
在上述色谱分析条件下,从同一个氟丙嘧草酯原药样品中准确称取6个试样,进行平行测定,考察方法的精密度,试验结果见表1。氟丙嘧草酯的标准偏差为0.11,变异系数为0.11%。结果表明该方法的重现性良好,精密度高,能够满足定量分析的要求。
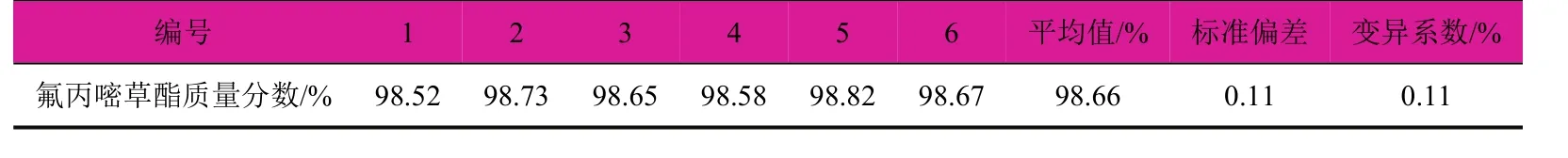
表1 精密度试验
2.5 方法准确度的测定
准确称取6个已知质量分数的氟丙嘧草酯原药样品,分别加入一定量的氟丙嘧草酯标准品,按照1.3章节的液相色谱条件测定待测样品中氟丙嘧草酯的质量,计算其回收率,结果见表2。氟丙嘧草酯的平均回收率为99.94%。试验结果表明该方法回收率较好,准确度高。
3 结 论
采用反相高效液相色谱法对氟丙嘧草酯原药进行了定性定量分析。试验结果表明:该方法简单快速,分离效果好,峰形尖锐对称,线性关系好,精密度和准确度高,适用于氟丙嘧草酯原药的定量分析,也为该产品相关制剂配方的分析研究提供了参考。
猜你喜欢
农药科学与管理(2022年6期)2022-08-03
系统医学(2022年6期)2022-06-13
云南农业科技(2022年1期)2022-01-27
中国食品(2020年16期)2020-08-31
——第二部分:原棉短纤维率标样的验证试验分析
中国纤检(2020年7期)2020-07-22
科学家(2016年17期)2017-10-17
今日农药(2017年7期)2017-08-09
科技视界(2016年22期)2016-10-18
今日农药(2014年12期)2015-03-30
科技视界(2015年15期)2015-01-16
