
基于JPPF的异构分布式MTH1虚拟筛选系统
2020-03-18 03:44陈云侠陈文波
计算机与现代化 2020年3期
陈云侠,张 洋,陈文波
(兰州大学网络安全与信息化办公室,甘肃 兰州 730000)
0 引 言
2014年在国际著名期刊《Nature》上报道了一种针对肿瘤癌细胞的激酶MTH1,文章指出MTH1是癌细胞生存所必需的,而在正常细胞中MTH1是非必需的[1-3]。MTH1是一种氧化嘌呤核苷三磷酸酶,内含抗氧化相关修复基因,能够将dNTP池中的氧化核苷酸进行选择地清除,为癌细胞生存所必须,所以对MTH1抑制剂的研究将对癌症的治疗有着重要的意义。
在药物研究领域中对于新药的发现工作主要使用虚拟筛选方法[4],这是一种计算密集型任务。在高性能计算的带动下这种方法有了突飞猛进的发展,极大地缩短了药物研究与开发的进程,提高了药物开发的效率。计算机辅助药物设计已经成为现代药物研发进程中不可替代的方法[5-6]。最近文献[7]用虚拟筛选方法来搜索潜在的药物用于治疗导致乙型脑炎的化合物NiV-G。文献[8]用计算模拟的方法预测出雌二醇可能是治疗CMT1A患者的首选药物,CMT1A在不久的将来可能会引起生物学家的更多关注。
网格是大规模虚拟筛选的首选技术,有报道称使用Worldwide Grid的网格计算资源成功筛选了抗甲型流感病毒的药物[9];文献[10]利用EELA-2网格研究了在委内瑞拉的登革热病毒的分子流行病学的机制;有学者专门设计了名叫Hydra的软件,使用165节点进行了基于网格计算的药物虚拟筛选的计算[11];开源的分布式计算软件Hadoop已经被用于基于UCSF Dock(仅限于LinuxOS)的药物虚拟筛选中[12]。但是,网格技术需要先进的架构,高效的工具、软件等来管理,才能最大化利用共享的资源和架构,成本非常高;其次,它严重依赖分散的数据管理,一些软件的定义不明确,有些应用须修改才能适应网格的使用,这些问题导致了网格技术比较难以进一步推广和应用[13]。Hadoop是MapReduce计算模型的一个实现框架,它采用了共享的HDFS文件系统。该文件系统对本地服务器的磁盘读写要求很高,且会在节点间产生大量的流量,故对网络的通畅性要求很高。所以,Hadoop架构只有在完成网络优化后,才有可能实现计算性能提升[14]。以虚拟化技术为主的云计算,面对以性能为主的高性能计算应用来说也不是一个很好的解决方案。
分布式计算在现代药物设计中起到越来越重要的作用[15-17]。虽然很多关于药物的分布式计算的研究已经陆续被报道,但是相关的源码和成型的软件尚没有被报道。对于一般的实验室来讲,组建灵活方便并且稳定有效的网格去进行药物的虚拟筛选仍然是一项不容易的工作。
JPPF提供了一种很好的异步分布式计算框架,可以管理和充分利用不同架构乃至不同操作系统下的计算资源。如果将这些资源组织起来并高效地利用,就可以进行大规模的虚拟筛选。不仅如此,通过JPPF(Java Parallel Processing Framework, Java并行处理框架)还可以统一调配后期的分子动力学模拟的任务,将其部署到计算能力更高的GPU计算平台上。GPU架构的机器可以加速分子动力学模拟的计算,实验测试中1台GPU就可以达到普通集群约224核心的计算能力[18]。所以,对于基于分子对接的虚拟筛选和分子动力学模拟来说,组建不同架构的平台资源进行调度和参与计算就可以满足不同方面的计算需要。
本文以JPPF异步分布式计算框架和AutodockVina分子对接技术作为基础技术,设计一种异步分布式虚拟筛选系统,通过对100万药物分子集进行筛选,最终得到MTH1抑制剂的目标结果集。
1 系统的主要技术简介
1.1 JPPF
JPPF是一个开源的网格计算架构,它可以在分布式环境下同时运行多个Java应用,利用它可以简单方便地构建异构分布式计算环境来处理大规模并行问题[19]。
为了减少处理时间,JPPF将一个应用分解成能够在多台计算机上同时执行的小任务来实现,使得需要大规模处理能力的应用可以在任意数量的计算机上运行。JPPF提供了多种负载平衡算法、故障转移、错误恢复和应用级安全策略,并可以与J2EE集成。JPPF具备下列特点:可以快速建立和运行JPPF网格;根据需求动态扩展;与领先的J2EE应用服务器连接;易于编程的模型;细粒度的监控和管理;容错和自我修复能力;高性能的服务水平和可靠性。
JPPF包括clients、servers和nodes这3个部分。clients通过调用JPPF API将应用程序的计算任务提交给JPPF网格;servers接收来自clients的任务并将任务分派到nodes上,然后接收从nodes计算得到的结果并将结果返回给clients;nodes执行真正的计算任务[20]。这3部分之间的关系如图1所示。
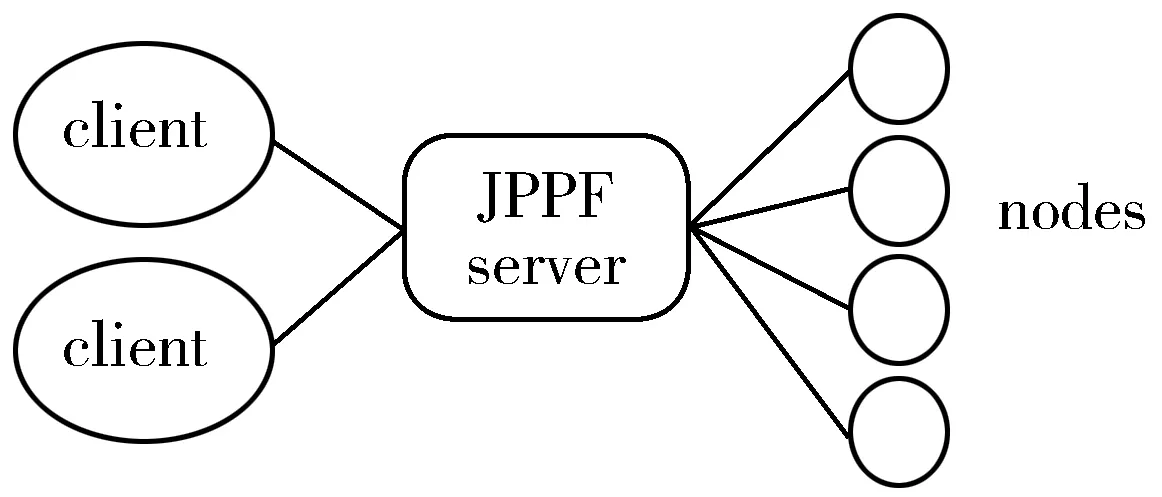
图1 JPPF的基本结构
由图1可以看出,server扮演着非常重要的角色,负责client和nodes之间的通信。在实际部署中,如果采用一个server负责所有处理情况,那么当server出现故障或崩溃时整个JPPF网格将瘫痪。JPPF可以扩展成包含多个server的结构,各server之间通过P2P的网络连接方式进行通信,并且对于clients和nodes提供了额外的连接选项和容错机制。在这种拓扑结构中,每一个server视它的对等节点为一个client,并被它的对等节点看作是一个node,这极大地增强了JPPF的可靠性。
1.2 AutoDockVina
AutoDockVina是一款由Molecular Graphics实验室的Dr. Oleg Trott开发的用于分子对接和虚拟筛选的开源软件[21]。目前该软件已经历2代,分别是AutoDock 4和AutoDockVina,其中AutoDock 4是AutoDockVina之前的一个版本。除了用于分子对接之外,这2个版本还可以实现原子之间引力关系的可视化,这可以指导有机合成化学家设计更好的所需材料[22]。AutoDockVina因为具有准确性高、运算速度快、容易使用等特征,所以与其他同类软件相比具有极强的竞争优势。相对于之前的版本AutoDock 4,AutoDockVina通过在多核机器上使用多线程技术以及充分利用所有的CPU和GPU资源,极大地提高了绑定模式预测的平均性能,实现了2个数量级的性能提升,进一步提高了绑定模式预测的精度[23]。此外,AutoDockVina已经严格地通过被称为Directory of Useful Decoys基准程序的测试。在兼容性方面,AutoDockVina与AutoDock 4都采用PDBQT分子结构文件格式,该文件可以通过MGLTools产生和查看,具有很好的兼容性。鉴于AutoDockVina的设计理念,用户并不需要理解其实现细节、搜索参数、聚合结果等,具有简单容易的特点。
2 系统设计与实现
2.1 系统架构
基于JPPF的虚拟筛选计算系统架构如图2所示,主要包括4个部分:Molgridcal、Server、计算节点和网络。

图2 虚拟筛选系统架构
Molgridcal主要完成对筛选数据的预处理,即从不同的药物数据库导入需要筛选的分子,对分子进行转化并建立最终筛选的数据库。该系统选取ZINC、Pubchem和SuperNature这3个药物数据库的分子。首先将ZINC数据库中的小分子直接导入基于ChemAxon和MySQL建立的数据库。对于Pubchem和SuperNature数据库中的2D分子,通过脚本和ChemAxon过滤掉与数据库中已经收录的重复结构的小分子,并将其转化成3D结构,最后将处理好的全部小分子都导入到指定的数据库中,以备虚拟筛选。Server主要是将待筛选的数据分配到相应的计算节点上。
该系统设计了2种方法用于系统的负载均衡:一种是了解每一个计算资源的计算能力,手动发送不同的任务量到不同的计算节点;另外一种是使用Monte Carlo的自适应启发式算法分配任务到不同的计算节点。计算节点主要由各种异构计算资源组成,用于对目标的任务进行虚拟筛选或者模拟。可靠的网络保证分子数据在虚拟筛选过程的有效传输。
2.2 实现过程
1)准备待筛选的100万目标分子数据,部署数据存储服务器。
2)部署和配置Molgridcal和Server。
3)启动Molgridcal,对待筛选的数据进行读取,等到Molgridcal对数据读取结束,启动Server。
4)当计算节点连接上相应的Server之后,此计算节点会从Molgridcal上下载一个待筛选的分子,然后进行计算。计算结束之后,将节点的计算结果上传到数据服务器。
5)所有的分子筛选结束之后,将计算的结果进行分析。
3 实验结果分析
3.1 实验环境
虚拟筛选需要用到的实验环境主要分3个部分。
第1部分是数据中心,存储了所有的数据。使用的是华硕KCMA-D8,其硬盘容量为1 TB,操作系统使用的是CentOS release 6.2。
第2部分为JPPF的服务器,使用的是IBM System x3550,操作系统为CentOS release 6.3。
第3部分为计算节点,包括一个GPU集群,由4个曙光GPU工作站组成,每个GPU工作站节点配置2 个Intel Xeon 5650 2.66 GHz 6核CPU处理器,4块NVIDIA Tesla C2050 GPU卡,使用的操作系统为CentOS release 5.4。一个CPU集群,包含3个Intel Xeon 5650服务器节点,每个节点拥有2个Intel Xeon 5650双核CPU 处理器,操作系统为CentOS release 6.3。2台迷你台式电脑,其中1台使用的是Intel 酷睿i5 3470四核CPU处理器,操作系统为Microsoft Windows 7 Professional,另一台使用的是CentOS release 6.6,Intel酷睿2双核处理器。
3.2 MTH1分子模型
MTH1的分子模型如图3所示。其中,条状图代表蛋白质,球状图代表钠离子,其余的棍状模型图代表水分子。
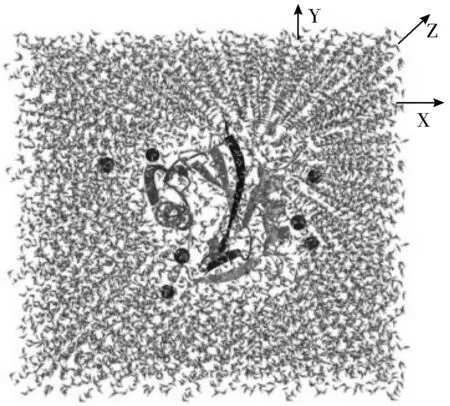
图3 MTH1的分子模型
3.3 100万MTH1类药分子划分和筛选
考虑到数据服务器的存储空间和内存限制,将100万的待筛选分子进行了划分并设置了每次筛选的分子数量,具体如表1所示。在每一个等待筛选的文件夹中放置40000个左右的分子,将任务分为25份。如果计算机的存储空间和内存足够大,可以设置每次筛选更多的分子。
表1 100万MTH1的类药分子的划分
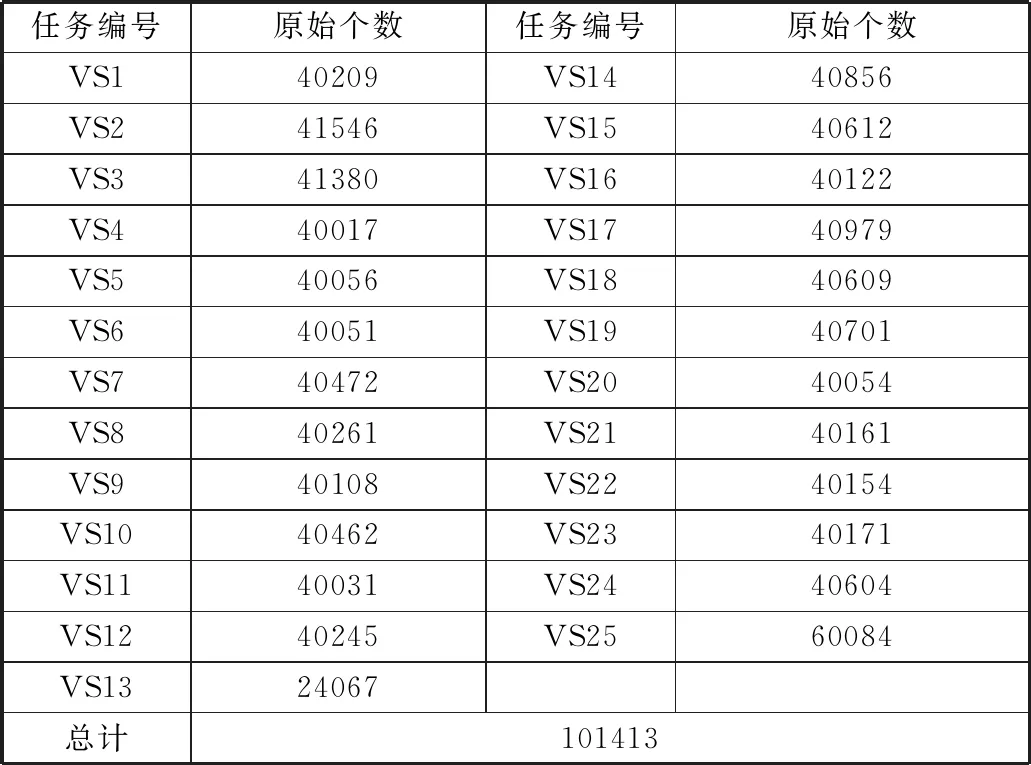
任务编号原始个数任务编号原始个数VS140209VS1440856VS241546VS1540612VS341380VS1640122VS440017VS1740979VS540056VS1840609VS640051VS1940701VS740472VS2040054VS840261VS2140161VS940108VS2240154VS1040462VS2340171VS1140031VS2440604VS1240245VS2560084VS1324067总计101413
以任务编号VS1的40209个待虚拟筛选分子为例,不同的计算资源(小型CPU集群、GPU集群、普通PC机)对其筛选情况如表2所示。
表2 不同计算资源的虚拟筛选情况(以VS1为例)
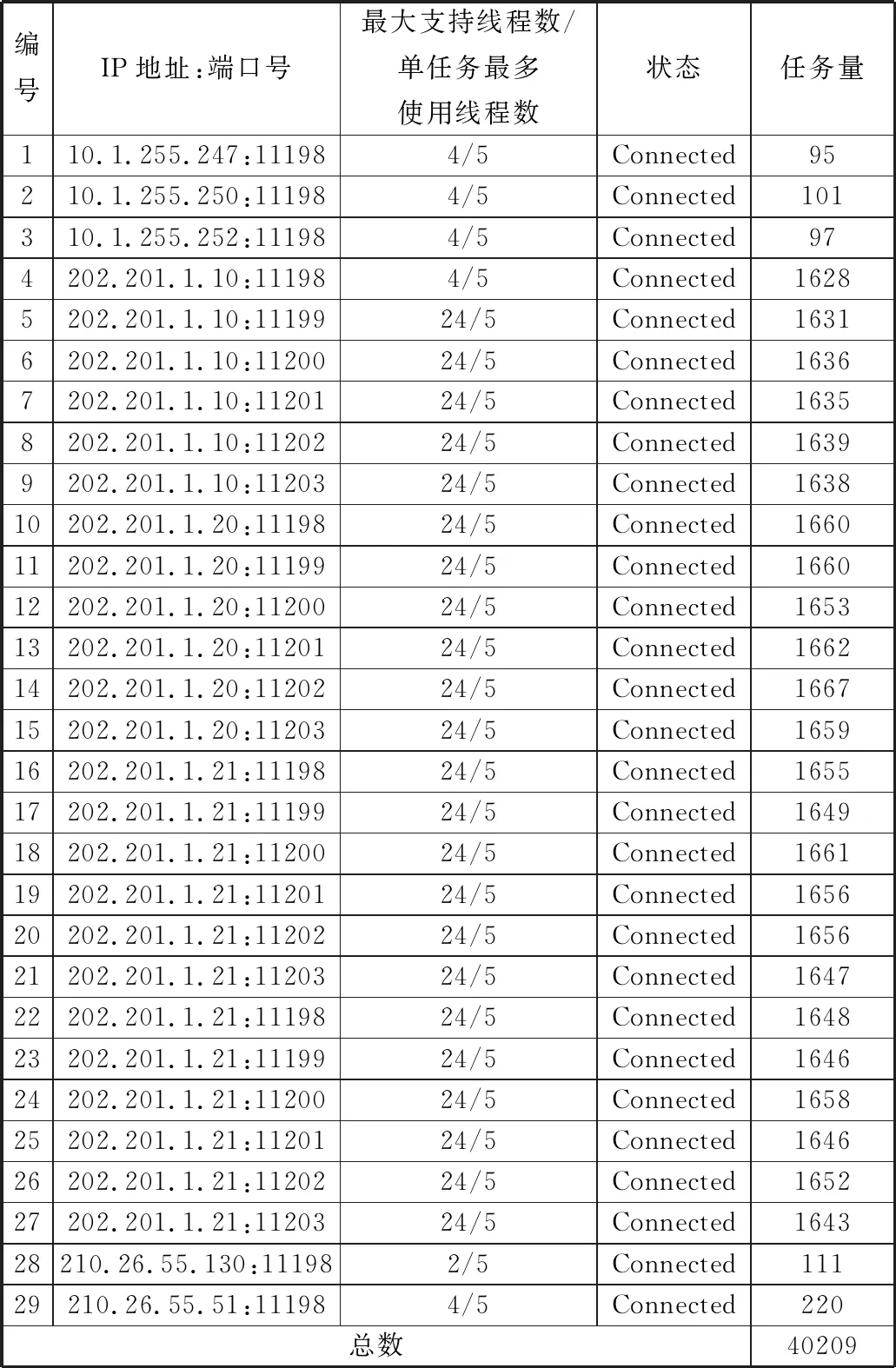
编号IP地址:端口号最大支持线程数/单任务最多使用线程数状态任务量110.1.255.247:111984/5Connected95210.1.255.250:111984/5Connected101310.1.255.252:111984/5Connected974202.201.1.10:111984/5Connected16285202.201.1.10:1119924/5Connected16316202.201.1.10:1120024/5Connected16367202.201.1.10:1120124/5Connected16358202.201.1.10:1120224/5Connected16399202.201.1.10:1120324/5Connected163810202.201.1.20:1119824/5Connected166011202.201.1.20:1119924/5Connected166012202.201.1.20:1120024/5Connected165313202.201.1.20:1120124/5Connected166214202.201.1.20:1120224/5Connected166715202.201.1.20:1120324/5Connected165916202.201.1.21:1119824/5Connected165517202.201.1.21:1119924/5Connected164918202.201.1.21:1120024/5Connected166119202.201.1.21:1120124/5Connected165620202.201.1.21:1120224/5Connected165621202.201.1.21:1120324/5Connected164722202.201.1.21:1119824/5Connected164823202.201.1.21:1119924/5Connected164624202.201.1.21:1120024/5Connected165825202.201.1.21:1120124/5Connected164626202.201.1.21:1120224/5Connected165227202.201.1.21:1120324/5Connected164328210.26.55.130:111982/5Connected11129210.26.55.51:111984/5Connected220总数40209
表2中,编号1~3为小型的CPU集群,每一个节点有4个CPU计算核心;编号4~27为GPU集群,有4个节点,由于每一个计算节点的CPU计算核心有24个,而每一个筛选任务最多只能使用5个核心,因此在一个节点上重复运行了6个任务;编号28~29为普通PC机,可以使用的CPU计算核心数分别为2个和4个。
由表2可以看出,所有的计算资源都能成功地连接,同时对相关的任务进行计算。由于GPU集群不仅拥有超强计算能力的GPU,同时还拥有较多的CPU计算核心,因此它的计算能力是最强的。在只是使用一个计算节点的情况下,GPU集群是CPU集群计算速度的990倍,是双核PC机的900倍,是4核PC机的450倍。因此,虽然CPU的计算能力和核数对计算速度有一定的影响,但是GPU对系统计算速度的提升更加明显和有效。
3.4 分析及结论
本次实验采用不同构架的计算资源实现了对MTH1类药分子的虚拟筛选,并筛选出前10名的类药分子,具体如表3所示。前10名类药分子用于进一步的分析,排名越靠前的分子说明是亲和力越强的分子。
表3 虚拟筛选出的前10名类药分子

编号类药分子亲和力/(kcal/mol)1ZINC72003306-13.12ZINC09559647-13.13ZINC13750217-13.04ZINC04141488-12.95ZINC63097139-12.96ZINC04815059-12.97ZINC04909555-12.98ZINC78511930-12.69ZINC16780432-12.610ZINC35882888-12.6
系统选取了MTH1晶体结构中的配体TH287作为对照,通过分子对接得到TH287和MTH1的亲和能是-8.7 kcal/mol。而通过虚拟筛选得到的前10名类药分子都是低于这个数值的,说明这些分子是潜在的MTH1的配体。
以排名第1(No.1)的类药分子(ZINC ID:ZINC72003306)为例,分子结构如图4所示。
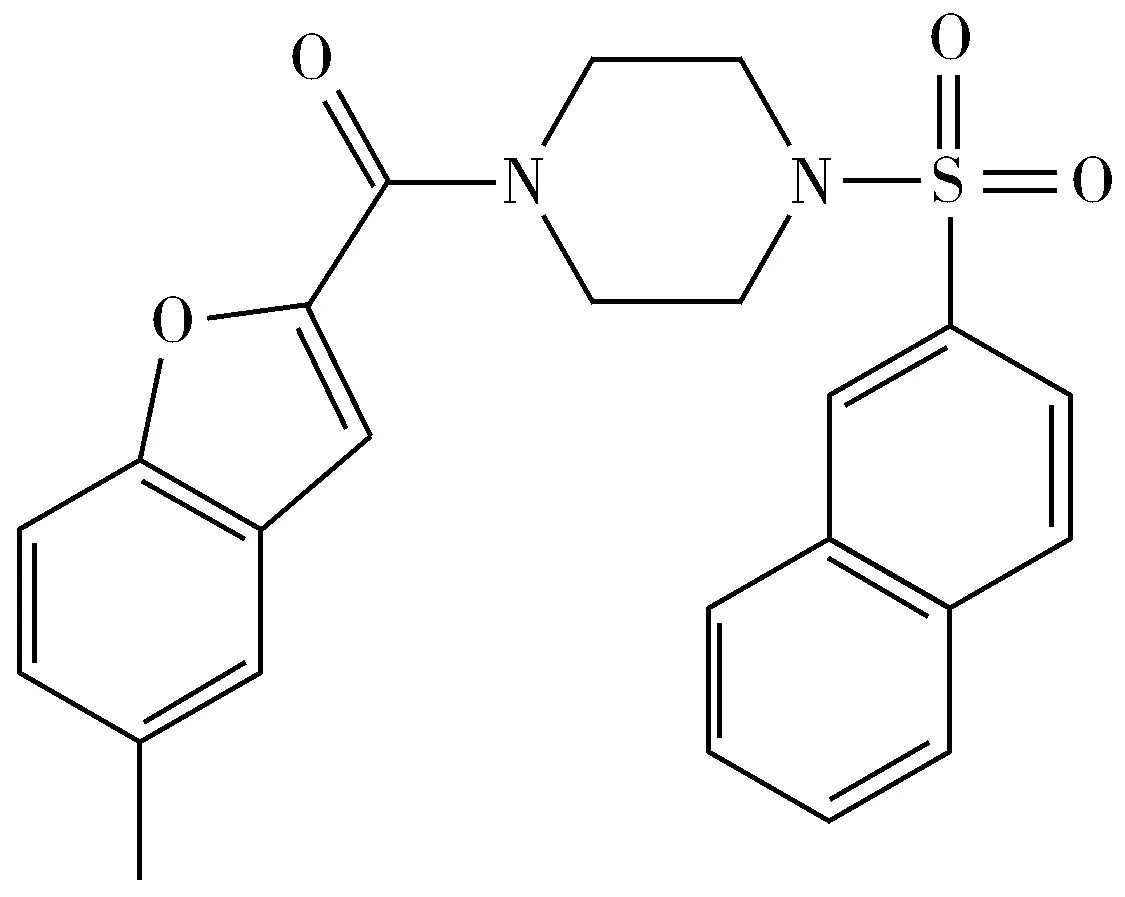
图4 ZINC72003306的分子结构
MTH1活性口袋中的活性位点由氨基酸N33、D119和D120组成。如图5所示,通过比较TH287和No.1分子在MTH1结合口袋中的结合模式可以看到,No.1分子和TH287都能结合在氨基酸N33、D119和D120形成的空腔周围,表明了通过虚拟筛选的方法可找到直接靶向MTH1的药物分子。
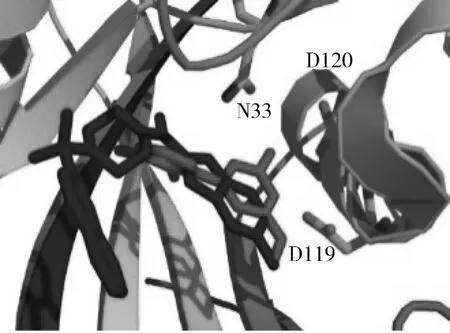
图5 TH287和类药分子ZINC72003306及MTH1的结合模式
4 结束语
虚拟筛选需要大规模的计算资源,但是传统的网格技术严重依赖分散、高效的数据管理,已经不适合应用到大规模的肿瘤药物筛选和模拟。而通过对分布式资源的弹性调度不仅可以集中计算资源,还可以整合可利用的空闲资源一起参与这种大规模分布式应用。同时,既能节约购买计算资源的成本,还可以充分发挥每一种资源的利用率,满足不同的计算需求。本文设计了一种基于JPPF分布式并行框架的虚拟筛选系统,该系统使用了不同类型的计算资源,包括普通PC机、CPU集群和GPU集群。为了测试JPPF药物筛选系统的性能,系统对100万药物分子集进行了筛选,并选出排名前10名的类药分子。通过比较TH287和No.1分子在MTH1结合口袋中的结合模式,发现No.1分子和TH287都能结合在氨基酸N33、D119和D120形成的空腔周围,表明使用本文设计的虚拟筛选方法可以找到直接靶向MTH1的药物分子。实验同时验证了不同计算资源的计算速度和计算能力成正比。该系统的实现为快速构建大规模药物分子虚拟筛选技术提供了一种解决方案。
猜你喜欢
科学技术创新(2021年18期)2021-06-23
微型电脑应用(2019年10期)2019-10-23
中国生殖健康(2019年9期)2019-01-07
计算机测量与控制(2017年12期)2018-01-05
计算机技术与发展(2017年12期)2017-12-20
软件导刊(2016年12期)2017-01-21
软件导刊(2016年11期)2016-12-22
科学与财富(2016年15期)2016-11-24
中国医院用药评价与分析(2014年8期)2014-01-23
中国药房(2013年10期)2013-12-03
