
胺脱氢酶催化合成手性胺的机遇与挑战
2020-03-12 02:51陈飞飞汪东浩许建和郑高伟
生物加工过程 2020年1期
陈飞飞,汪东浩,许建和,郑高伟
(华东理工大学生物反应器工程国家重点实验室,上海200237)
手性胺广泛存在于各种生物活性分子中,是合成药物、天然产物、农药、精细化学品等产品的重要手性中间体。近年来,美国FDA批准上市的化学药物分子中约40%含有手性胺的结构(图1),正所谓“无氮不成药”[1]。此外,手性胺还被作为手性拆分剂、手性配体等用于各种手性化合物的制备。手性胺的合成已经引起学术界和工业界的广泛关注,其中酮的不对称还原胺化,在2007年由美国化学会与辉瑞等十余家全球制药巨头企业联合召开的圆桌会议上,被认定为制药工业中第二理想的反应[2],这是因为酮的不对称还原胺化制备手性胺具有只生成副产物H2O、使用廉价的氨作为氨基供体、产品分离过程简单等优势。然而大多数化学合成途径需要高温高压等苛刻的反应条件、使用有毒有害的试剂和重金属催化剂、多步结晶提升产品的光学纯度,这些限制了它们在工业中的广泛应用[3]。因此亟待研究开发更加绿色的途径替代这些不可持续的生产工艺。

图1 含有手性胺结构的药物分子Fig.1 Pharmaceuticals contain a chiral amine moiety
随着定向进化等蛋白质工程技术的发展,酶催化已成为替代药品、精细化学品和农用化学品等化学合成途径的重要手段,特别是对于手性化学品的合成[4-6],这主要是因为它具有立体选择性高、反应条件温和、环境友好等优点。已报道的用于合成手性胺的酶主要有脂肪酶[7]、转氨酶[8-10]、胺氧化酶[11-13]、氨裂解酶[14-15]、亚胺还原酶[16-19]和还原胺化酶[20]等。其中,近年来开发的胺脱氢酶(amine dehydrogenase,AmDH)为手性胺的合成提供了更理想的合成途径。它能够以NAD(P)H为辅因子催化酮与氨发生不对称还原胺化反应,合成相应的手性胺,该过程使用比较廉价的氨作为氨基供体,所生成的副产物只有H2O,因此被认为是更加绿色的合成途径(图2)。

图2 胺脱氢酶催化酮不对称还原胺化反应Fig.2 Asymmetric reductive amination of ketones to chiral amines by amine dehydrogenases
自从2012年美国佐治亚理工学院的Abrahamson等[21]首次通过分子进化创制出工程胺脱氢酶以来,对胺脱氢酶的研究引起了全球的广泛关注。目前已有多个胺脱氢酶被报道(表1),不仅包括开发出的多种工程胺脱氢酶,也含有鉴定出的天然胺脱氢酶。通过蛋白质工程技术拓展了酶的底物谱,利用固定化、反应介质设计等传统手段强化了酶的催化效率和生产强度,特别是利用新开发的胺脱氢酶,设计构建一系列多酶级联反应途径,为从廉价原料到高附加值手性胺的高效简洁合成提供了可行性。本文中,笔者主要综述胺脱氢酶近些年来取得的一些重要研究进展,并预测其未来面临的发展机遇与挑战。

表1 文献报道的胺脱氢酶
1 天然胺脱氢酶的发现
1999年,日本富山县立大学的Itoh等[22]首次从自然界中鉴定出了一个来源于Streptomycesvirginiae的NADH依赖型胺脱氢酶SvAmDH。该酶具有广泛的催化底物范围,可催化醛类、酮类、酮醇类及酮酸类化合物的还原胺化及甲胺、正己烷、丝氨醇等胺类化合物的氧化脱氨反应(图3),但是其存在活力较低、对映体选择性较差等缺点,且其核酸和蛋白质序列信息至今尚未报道。

图3 SvAmDH催化的反应类型Fig.3 Reductive amination reactions catalyzed by SvAmDH
2016年,Vergne-Vaxelaire课题组的Mayol等[23]通过基因挖掘和对构建的酶库进行功能筛选,获得了能够催化4-氧代戊酸不对称还原胺化生成(S)-4-氨基戊酸的天然胺脱氢酶AmDH4,其对4-氧代戊酸的催化比酶活可达50 mU/mg,且在60 ℃下的半衰期长达65 h。该酶可以高效催化高达500 mmol/L的4-氧代戊酸不对称还原胺化生成(S)-4-氨基戊酸,产品得率达88%,光学纯度(对映体过量值e.e.)达99.5%以上(图4),表现出了极好的应用潜力。

图4 胺脱氢酶催化的4-氧代戊酸不对称还原胺化反应Fig.4 Reductive amination of 4-oxopentanoic acid to (S)-4-aminopentanoic acid catalyzed by AmDH4
在上述研究的基础上,最近Vergne-Vaxelaire课题组的Mayol等[30]通过进一步基因挖掘得到一系列可催化酮不对称还原胺化的天然胺脱氢酶(nat-AmDH)。该家族的胺脱氢酶与之前报道的催化还原胺化反应的酶如工程胺脱氢酶、亚胺还原酶和还原胺化酶均非同一家族。为探明该家族胺脱氢酶的结构与功能关系,他们解析了3种代表性酶的晶体结构(图5)。在此基础上,对AmDH4底物结合口袋附近的极性氨基酸突变为疏水性氨基酸,最终构建的四点组合突变体AmDH4N135V/N163V/R161M/H264L对原本难以催化的疏水型底物2-戊酮表现出良好的催化性能,比酶活可达104.8 mU/mg。此外,对此类天然胺脱氢酶家族2 000多条序列进行了分析,揭示了该家族酶系的进化与聚类关系。此酶家族分为5个亚家族,G1、G2、G3和G4进化关系相对较近,其中G1亚家族的酶不具有2,4-二氨基庚二酸脱氢酶的活性,而G2亚家族的酶(包含AmDH4)具有此种活性。G3和G4亚家族中均发掘出天然胺脱氢酶,G5亚家族的进化关系相对其他4个亚家族较远。此研究为后续此类胺脱氢酶的进化与应用研究提供了一定的研究基础。

(a)AmDH4与辅酶NAD+的结构 (b) CfusAmDH与辅酶NADP+的结构 (c)MsmeAmDH与辅酶NADP+的结构图5 3种nat-AmDH家族胺脱氢酶的晶体结构Fig.5 Structures of three native amine dehydrogenases
2 工程胺脱氢酶的开发
2.1 基于亮氨酸脱氢酶改造的胺脱氢酶
氨基酸脱氢酶(amino acid dehydrogenase,AADH)是一类以NAD(P)H为辅因子的氧化还原酶,它能够可逆地催化氨基酸的氧化脱氨和α-酮酸的不对称还原胺化。2012年,Abrahamson等[21]以天然存在的Bacillusstereothermophilus亮氨酸脱氢酶为模板,通过多轮的分子进化,首次创制出了新颖的非天然胺脱氢酶,将酶活性口袋中与天然底物羧基结合的2个关键位点K68和N261由亲水性残基突变为疏水性残基68S和261L之后,其催化的底物类型由脂肪族氨基酸改变为不含羧基的脂肪酮类化合物(图6),这也表明K68和N261是以氨基酸脱氢酶为模板创制工程胺脱氢酶的关键突变位点。
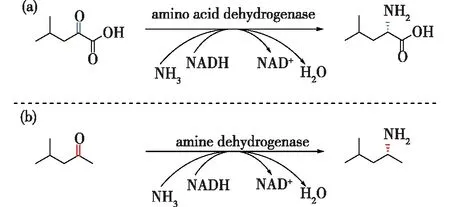
图6 天然氨基酸脱氢酶(a)与工程胺脱氢酶(b) 催化的反应Fig.6 Asymmetric reductive amination of keto acid by amino acid dehydrogenase (a) and ketone by amine dehydrogenase (b)
2.2 基于苯丙氨酸脱氢酶改造的胺脱氢酶
2013年,Abrahamson等[24]又以来源于Bacillusbadius的苯丙氨酸脱氢酶结构为进化模板,对与羧基结合的两个关键残基K77和N276进行组合饱和突变,通过筛选获得了底物谱改变的突变体K77S/N276L,其催化的底物由苯丙氨酸转变为无羧基的苯基丙酮类似物,从而创制出了苯丙胺类胺脱氢酶,它能够催化芳香酮的不对称还原胺化,与通过亮氨酸脱氢酶改造而来的胺脱氢酶,在底物谱上形成了互补,弥补了亮氨酸脱氢酶来源的胺脱氢酶难以催化芳香酮化合物还原胺化的不足。将该酶应用于芳香酮底物4-氟苯基丙酮的不对称还原胺化,合成了(R)-4-氟-α-甲基苯乙胺,产品e.e.值高达99.8% (图7),进一步证明通过对氨基酸脱氢酶的分子进化创制胺脱氢酶是可行的。随后,该课题组将亮氨酸脱氢酶和苯丙氨酸脱氢酶进化而来的两种胺脱氢酶通过domain shuffling构建成了一个“杂合”的胺脱氢酶,该酶表现出与原来两种胺脱氢酶均不同的底物特异性[25]。

图7 胺脱氢酶催化的4-氟苯基丙酮不对称还原胺化Fig.7 AmDH-catalyzed asymmetric reductive amination of p-fluorophenylacetone
受此结果启发,随后多个课题组陆续报道了通过分子改造苯丙氨酸脱氢酶创制苯丙胺类胺脱氢酶的成功案例。例如,新加坡国立大学的Li课题组的Ye等[26]通过定向进化将来源于Rhodococcussp.M4的苯丙氨酸脱氢酶突变为苯丙胺类胺脱氢酶TM-pheDH,该酶不仅能够以4-氟苯基丙酮为底物,还能催化4-苯基-2-丁酮的不对称还原胺化(图8),进一步拓宽了苯丙胺类胺脱氢酶的催化底物谱。2017年,Schell课题组的Pushpanath等[27]也通过对嗜热菌Caldalkalibacillusthermarum来源的苯丙氨酸脱氢酶进行进化,获得了一个热稳定好(Tm83.5 ℃)的胺脱氢酶Cal-AmDH,该酶对苯丙酮类似物如4-氟苯基丙酮、2-氟苯基丙酮、4-甲基苯基丙酮及苯氧基丙酮等底物均显示出较高的催化活力,并且在两相反应体系中实现了高达400 mmol/L苯氧基丙酮的不对称还原胺化,表现出了一定的实用前景。
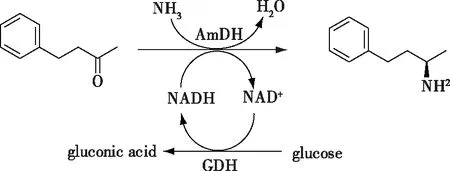
图8 胺脱氢酶TM-pheDH催化的4-苯基-2-丁酮 不对称还原胺化Fig.8 Asymmetric reductive amination of 4-phenyl- 2-butanone catalyzed by amine dehydroge- nase TM-pheDH
2.3 基于L-赖氨酸ε-脱氢酶改造的胺脱氢酶
最近,荷兰阿姆斯特丹大学的Mutti课题组的Tseliou等[28]以L-赖氨酸ε-脱氢酶作为模板,对活性位点周围残基进行突变,获得了具有不对称还原胺化功能的胺脱氢酶(LE-AmDH-v1),该酶不仅具有非常高的热稳定性,而且具有较宽的底物谱,对醛酸、酮酸、醛、脂肪酮和芳香酮等化合物都显示出一定的胺化活性。
2.4 胺脱氢酶的分子改造
工程胺脱氢酶都是由天然氨基酸脱氢酶改造而来的,它们能催化的化合物都是天然底物的类似物,存在底物谱偏窄的问题,这极大地限制了它们在手性胺合成中的应用范围。例如,由亮氨酸脱氢酶改造而来的胺脱氢酶,其底物脂肪酮的链长通常小于6个碳原子。笔者所在课题组[29]利用自主研发的氨基酸脱氢酶作为模板,通过生物分子工程手段开发出了3个新的胺脱氢酶,并针对其底物谱较窄的问题,采用计算机辅助的蛋白质工程技术,确定了酶活性口袋中影响酶与大位阻底物结合的关键残基(Ala113与Thr134),通过将其突变成分子最小的甘氨酸(Gly),所构建的突变体LfAmDH(A113G/T134G)实现了对活性口袋的“容积拓展”(图9),从而将该酶所催化的底物范围由最长6个碳链长的脂肪酮拓展至长达10个碳链长的脂肪酮(如2-庚酮、2-辛酮和2-壬酮),显著拓宽了该酶催化的底物范围。此外,这两个关键氨基酸残基的突变在另外2个同源胺脱氢酶的改造中也具有类似的效果,这为其他不同胺脱氢酶的底物谱拓展提供了有益参考。

图9 胺脱氢酶LfAmDH与其突变体的底物结合口袋对比Fig.9 Illustration of the substrate-binding pockets of LfAmDH and its mutant (A113G/T134G) with docked substrate 2-heptanone
最近,笔者所在课题组[31]又对来源于Lysinibacillusfusiformis的氨基酸脱氢酶进行分子改造,创制出了能够催化α-羟基酮不对称还原胺化的胺脱氢酶(图10),并通过进一步的结构改造,拓展了底物结合口袋的空间,拓宽了其催化的底物范围。最优突变体可以高立体选择性地不对称还原胺化不同链长α-羟基酮,合成相应的(S)-邻位氨基醇,该酶也已成功用于抗结核药乙胺丁醇前体的合成。该工作将胺脱氢酶的底物谱由酮类化合物拓展到羟基酮类化合物,为手性药物中间体邻位氨基醇的合成提供了新颖的生物催化剂。

图10 胺脱氢酶催化的α-羟基酮不对称 还原胺化合成手性邻位氨基醇Fig.10 Asymmetric reductive amination of α-hydroxy ketones catalyzed by amine dehydrogenase for chiral vicinal amino alcohols synthesis
3 反应工程调控
3.1 两相反应体系
开发有机相/水相两相反应体系是生物催化的一种重要策略,可以解决底物或产物对于酶的动力学抑制作用,或者在热力学上促进反应向着产物生成的方向进行。胺脱氢酶催化的大多数底物都是水不溶性的羰基化合物,导致较低的生产效率,而常规添加助溶剂的策略经常导致胺脱氢酶的失活。为了解决这一问题,Au等[32]开发了庚烷/缓冲液两相介质反应体系(图11),显著提升了手性胺的合成效率,也实现了对在水相中水相溶解性差底物(如1-金刚烷基甲基甲酮等)的不对称还原胺化。

图11 胺脱氢酶两相反应体系实现疏水性酮 底物的不对称胺化还原Fig.11 A biphasic reaction system for asymmetric reductive amination of hydrophobic ketone substrates using AmDH
Pushpanath等[27]针对产物对胺脱氢酶存在抑制的问题,也开发了乙酸异戊酯/铵盐缓冲液双相反应体系,有效解决了高浓度底物/产物导致的胺脱氢酶Cal-AmDH失活问题,实现了高达400 mmol/L苯氧基丙酮的高效生物转化,产生的(R)-1-苯氧基-2-丙胺的e.e.值达99%以上。
由此可见,反应介质的设计也能有效提升胺脱氢酶高效合成手性胺的效率,未来研究中可以尝试其他反应介质,如反胶束、离子液体等。
3.2 酶的固定化
酶的固定化是提升酶稳定性的常用策略,有利于酶的回收及循环利用。胺脱氢酶的固定化研究也已经被报道,如Liu等[33]成功实现了胺脱氢酶在磁性纳米颗粒上的固定化,他们将含有组氨酸标签的重组蛋白与镍-次氮基三乙酸功能化的纳米颗粒通过亲和吸附实现固定化,利用共固定化的胺脱氢酶与葡萄糖脱氢酶作为催化剂,实现了40 mmol/L 4-苯基-2-丁酮的不对称还原胺化,底物转化率达74%,产物e.e.值达99%。2016年,Ren等[34]报道了利用TiO2纳米颗粒对胺脱氢酶进行固定化,该研究首先利用聚乙烯亚胺包裹胺脱氢酶为酶创造亲水的微环境,再利用处理后的酶作为模板诱导钛前体物质的水解和凝缩,从而形成纳米固定化酶颗粒,显著提升了酶的热稳定性。此外,在实际的应用中,酶的固定化不仅有利于酶的回收再利用,也可以降低生产成本。因此,胺脱氢酶固定化技术的探索将会促进该酶在实际生产中的应用。
4 多酶级联反应
4.1 胺脱氢酶/醇脱氢酶催化的双酶“借氢”级联
醇的不对称胺化是化学合成中的重要路线,但是存在选择性差、使用过渡金属等问题[35]。2015年,Turner课题组的Mutti等[36]利用胺脱氢酶与醇脱氢酶(ADH)耦联构建了催化醇转化为手性胺的双酶“借氢”级联途径(图12)。该级联反应途径首先利用醇脱氢酶(来源于Aromatoleumsp.、Lactobacillussp.、Bacillussp.等)实现芳香族或脂肪族伯醇或外消旋仲醇的氧化,生成相应的醛或甲基酮中间产物,再通过第二步胺脱氢酶催化的胺化还原反应,将中间产物转化为胺产物。第一步氧化反应产生的还原性氢(NADH)可以被第二步胺化反应所利用,因此该级联反应是一个氧化还原力自给自足的过程,通过“借氢”实现醇到胺的转化,是一个原子经济性高的绿色转化过程。利用该级联反应将伯醇转化为相应胺产物的转化率高达99%;将仲醇转化为相应R型手性胺产物的转化率高达96%,e.e.值高达99%。

图12 由醇到胺的双酶“借氢”级联反应路径Fig.12 A two-enzyme “hydrogen-borrowing” cascade for amination of alcohols
几乎同时,笔者所在课题组[37-38]同样报道了胺脱氢酶与醇脱氢酶耦联实现外消旋仲醇到手性胺转化的相同研究思路,利用来源于Exiguobacteriumsibiricum的亮氨酸脱氢酶自主改造而来的胺脱氢酶EsAmDH与通过基因挖掘获取的来源于Streptomycescoelicolor的一株醇脱氢酶ScCR分别实现醇的氧化和随后的胺化过程,将一系列脂肪族仲醇和苯乙醇转化为相应的手性胺产物,胺的得率最高达97%,手性胺的e.e.值>99%。值得一提的是,相对于Turner课题组的报道,笔者独立开发的该级联路径所使用的醇脱氢酶ScCR对于外消旋仲醇的对映选择性较弱,因此可以将外消旋仲醇中的两种异构体同时氧化,避免了同时添加两种对映选择性互补的醇脱氢酶。另外,该级联反应路径无需对两步所用的酶进行纯化,使用粗酶粉可以实现较高的转化效率,简化了反应操作过程。在笔者课题组和Turner课题组提出此双酶级联概念基础上,不同课题组通过对醇脱氢酶辅酶依赖性的改造[39]、使用固定化酶技术[40]或采用全细胞催化[41-42],使该级联体系的应用性研究得以拓展。
4.2 P450/醇脱氢酶/胺脱氢酶级联催化碳氢键胺化
对烷烃化合物进行C—H键不对称胺化是合成手性胺的一条新颖的路径,但是化学法实现该路径面临着反应条件苛刻、使用过渡金属催化剂、选择性差等问题。2015年,Both等[43]实现C—H键胺化的全细胞催化体系,通过利用P450单加氧酶/醇脱氢酶/转氨酶三酶共表达的全细胞作为催化剂,实现了乙苯及其几种衍生物的C—H键胺化(e.e.值97.5%)。鉴于胺脱氢酶催化的胺化还原过程具有较高的原子经济性,笔者所在课题组[44]构建了P450单加氧酶/醇脱氢酶/胺脱氢酶耦联的级联的反应路径(图13),该级联反应路径同样利用P450单加氧酶催化烷烃的羟化,再利用醇脱氢酶ScCR和胺脱氢酶EsAmDH分别实现醇的脱氢和酮的胺化。利用构建的P450单加氧酶、木糖脱氢酶(NADPH依赖性,用于与P450单加氧酶实现辅酶循环)、醇脱氢酶、胺脱氢酶共表达的全细胞,分别实现了2种烷烃化合物(环己烷、乙苯)的C—H键胺化,其中环己胺的浓度14.9 mmol/L,(R)-α-氨基苯乙胺的浓度2.2 mmol/L (e.e.值>99%)。
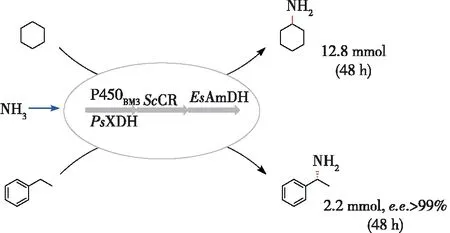
图13 多酶催化烷烃C—H键胺化的级联反应路径Fig.13 A multi-enzymatic cascade for C-H amination of alkanes
4.3 甲酸铵驱动的三酶级联反应催化酮R-选择性胺化
R选择性ω-转氨酶一般需要价格昂贵的D-丙氨酸作为氨基供体,而且存在反应平衡的问题,需要去除体系中产生的副产物以实现反应平衡向胺产物生成的方向移动[45-46]。为解决上述问题,笔者所在课题组[47]开发了ω-转氨酶/胺脱氢酶/甲酸脱氢酶三酶耦联的级联反应体系(图14)。在该体系中,甲酸脱氢酶消耗溶液中的甲酸根产生还原力NADH,胺脱氢酶可以利用溶液中的氨分子和还原力NADH将辅底物酮(如2-戊酮、2-己酮等)胺化为相应的胺产物,接着,R-选择性的ω-转氨酶使用上述产生的胺产物作为氨基供体实现目标酮底物的胺化。利用该反应系统,可以实现一系列酮底物(如4-苯基-2-丁酮、西他列汀前手性酮等)的R型胺化,转化率高达99%,e.e.值>99%。
因此,上述多酶级联体系可以利用甲酸铵作为氨基供体和还原剂,即可实现R-选择性的ω-转氨酶介导的R型胺化过程,且副产物仅为H2O和CO2,是一个简易、经济、绿色的R型生物催化胺化体系。

图14 甲酸铵驱动的三酶级联反应路径实现酮的R型胺化Fig.14 An ammonium formate driven trienzymatic cascade for ω-transaminase mediated R-amination
4.4 胺脱氢酶/转氨酶级联催化外消旋胺拆分
通过对外消旋体进行不对称拆分是制备特定对映体的常用生物催化策略。最近,韩国建国大学的Yoon等[48]构建了胺脱氢酶和S选择性ω-转氨酶级联的级联反应系统(图15),通过控制胺脱氢酶和S选择性ω-转氨酶的反应方向,可以分别实现R-型和S-型手性胺对映体的合成,分析得率高达100%,e.e.值>99%。利用该级联反应体系分别实现了(R)-2-庚胺和(S)-2-庚胺的制备级合成,分离得率分别为53%和75%,证明了该体系在手性胺制备合成中的实用性。此外,该课题组还构建了胺脱氢酶和丙氨酸脱氢酶耦联的级联路径[49],通过胺脱氢酶对外消旋胺进行氧化拆分可以实现S-型手性胺的制备,在该体系中,丙氨酸脱氢酶对丙酮酸进行还原胺化以实现NAD+的循环再生。

图15 胺脱氢酶/ω-转氨酶级联拆分 外消旋胺合成光学纯手性胺Fig.15 AmDH/ω-transaminase cascade systems catalyzing deracemization of racemic amines to chiral amines
5 结论与展望
近年来,由于胺脱氢酶可以利用廉价的氨水作为胺供体催化酮不对称还原胺化合成手性胺,对其研究引起了国内外学者的极大兴趣,也取得了显著的进展。但是相对转氨酶、脂肪酶、胺氧化酶等合成手性胺的酶系,对于该类酶的研究仍处于初始阶段,目前胺脱氢酶的种类还很少,高活性的酶还未见报道。因此,未来在新酶的开发、酶的催化性能提升、酶的结构-功能关系等方面还值得深入研究和探索,此外,该类酶也存在着极大的挑战,如其底物谱较窄、活性较低,如何突破这些限制,实现其在工业上的广泛应用,是值得深入研究的。总之,未来的研究重点应该是开发满足工业应用环境的胺脱氢酶催化剂,加速其在工业上的应用。
猜你喜欢
分子催化(2022年1期)2022-11-02
核安全(2022年3期)2022-06-29
功能材料(2022年5期)2022-06-02
家庭科学·新健康(2021年5期)2021-06-21
科学大众(2021年10期)2021-05-20
山西化工(2020年2期)2020-02-16
—— “T”级联
同位素(2019年1期)2019-03-14
心肺血管病杂志(2018年11期)2018-12-18
中国医药生物技术(2015年4期)2015-12-26
原子能科学技术(2015年12期)2015-07-07
