
N掺杂石墨烯负载Pt-Sn催化剂的制备及电催化性能研究
2020-03-06 01:52:24李慧君武宏钰王晓敏
化学工业与工程 2020年1期
李慧君,武宏钰,王晓敏,2*
(1.太原理工大学材料科学与工程学院,太原 030024; 2.新能源材料及器件山西省重点实验室,太原 030024)
直接乙醇燃料电池(direct ethanol fuel cells, DEFCs)具有高的能量密度和能量转化效率,无污染以及操作温度低等优点,在便携式电源方面有广泛的应用前景[1-3]。由于电极催化剂是燃料电池中重要的一部分,Pt催化剂一直是研究热点。但是Pt的抗毒性差、在载体表面易团聚、成本高等问题,限制了DEFCs的商业化应用[4-6]。现解决的主要途径是发展新型载体材料以提高催化效率,以及用 Pt与其他过渡金属形成合金来发展Pt基合金催化剂。
石墨烯是由sp2杂化的碳原子构成的单层蜂窝状二维网格结构的材料,具有高比表面积、良好的导电性、较高化学稳定性和突出的机械性等特点,被认为是DEFCs理想的电极催化剂载体材料。研究发现,经过氮原子掺杂的石墨烯(G-N)除具有以上特点外,其作为催化剂载体材料负载过渡金属催化剂对氧还原反应具有更好的催化活性和抗CO中毒性,近年来成为研究的热点。掺杂的N原子可以改变碳材料的电负性, 使得氮原子周围的碳原子带有更多的正电荷, 有利于氧气的吸附活化, 进而促进氧气的还原;掺杂的N原子也会影响C原子的自旋密度,当C原子的自旋密度较高时,就可作为氧还原反应的活性位点,当自旋密度是负值或者很小时,原子则具有更高的电荷密度,也可以作为活性位点,从而使石墨烯表面产生很多活性位点[7-8]。前期大量Pt/G-N、Pt-X/G-N 催化剂制备的文献报道表明[9-10],其相比Pt/G、Pt-X/G催化剂,催化性能明显提高。另一方面,为了降低成本、提高Pt的抗CO中毒能力,用Pt和其他过渡金属形成的合金代替Pt作为DEFCs的催化剂,研究趋向于Pt基合金催化剂,例如Pt-Au、Pt-Co、Pt-Sn和Pt-Ni等,其中Pt-Sn合金由于其良好的协同效应更具有一定的实际研究价值。
本课题组在前期工作中,通过多元醇法制备出了一系列Pt-Sn/石墨烯双金属复合催化剂,分别研究了Sn含量[11]、水含量[12]和pH值[13]对乙醇电催化性能的影响。在此基础上,本实验选用水合肼作为氮源,并作为还原剂还原GO,来制备掺氮石墨烯(G-N),这个方法简单而又不会在掺氮过程中再次造成石墨烯的结构缺陷。再使用乙二醇将H2PtCl6和SnCl2还原后的Pt-Sn纳米颗粒负载在经不同量的水合肼还原制得的G-N上。通过XRD、XPS、SEM和TEM等分析手段对样品进行结构和成分分析,并结合电化学分析,对比研究不同水合肼还原对催化剂结构、成分及性能的影响。
1 实验
1.1 催化剂的制备
通过改进的Hummers制备氧化石墨烯(GO),把获得的GO超声分散于去离子水中配成浓度为0.75 g/L的溶液;再加入不同质量的水合肼,GO与水合肼的质量配比是1∶3、1∶5、1∶7和1∶9。将混合溶液超声1 h后,在N2气流下磁力搅拌并加热到100 ℃保温24 h;之后将4 mL H2PtCl6溶液(0.050 mmol/L)、4 mL SnCl2溶液(0.025 mmol/L)和80 mL乙二醇溶液加入上述冷却到室温的混合液中,超声处理1 h;然后利用氢氧化钠乙二醇饱和溶液调整混合液的pH值大于12,并在N2气流下加热到130 ℃保温3 h;冷却到室温,用浓HNO3调整pH值小于2,继续搅拌20 h后,使用乙醇和去离子水分别对其进行离心洗涤,然后在40 ℃下真空干燥得到一系列不同的Pt-Sn/G-N催化剂。与其作对比的Pt-Sn/G使用乙二醇在相同条件下还原GO制备其载体材料石墨烯并负载Pt-Sn。
1.2 结构、成分和形貌表征
采用TD-3500型X-射线衍射仪(XRD,辐射源为Cu_Kα,管电压为30 kV,管电流为20 mA)测试样品的晶体结构;采用FTS-165型傅里叶红外光谱仪(波长4 000~400 cm-1)分析样品的各种基团;采用ESCALAB250型X射线光电子能谱仪(XPS,激发源为单色化Mg靶)分析样品表面的成分、价态及其含量;采用JEM-2010型高分辨透射电子显微镜(HR-TEM,电子枪为LaB6,加速电压为200 kV)观察样品的表面形貌及其金属纳米颗粒的分布。
1.3 电化学测试
电化学性能测试使用上海辰华CHI660D电化学工作站。利用传统的三电极测试方法,饱和甘汞电极为参比电极,铂丝为对电极,工作电极是直径4 mm的玻碳电极。催化剂溶液的配置:称取5 mg催化剂分散在1 mL的异丙醇和10 μL 的5%的Nafion混合溶液中并超声30 min。使用微量进样器量取3 μL的催化剂溶液均匀涂在电极表面,室温下放置干燥后进行测试。电解液在电化学测试前通入30 min N2除去空气。
2 结果和讨论
2.1 结构与成分分析
图1为不同Pt-Sn/G-N催化剂的XRD图谱。
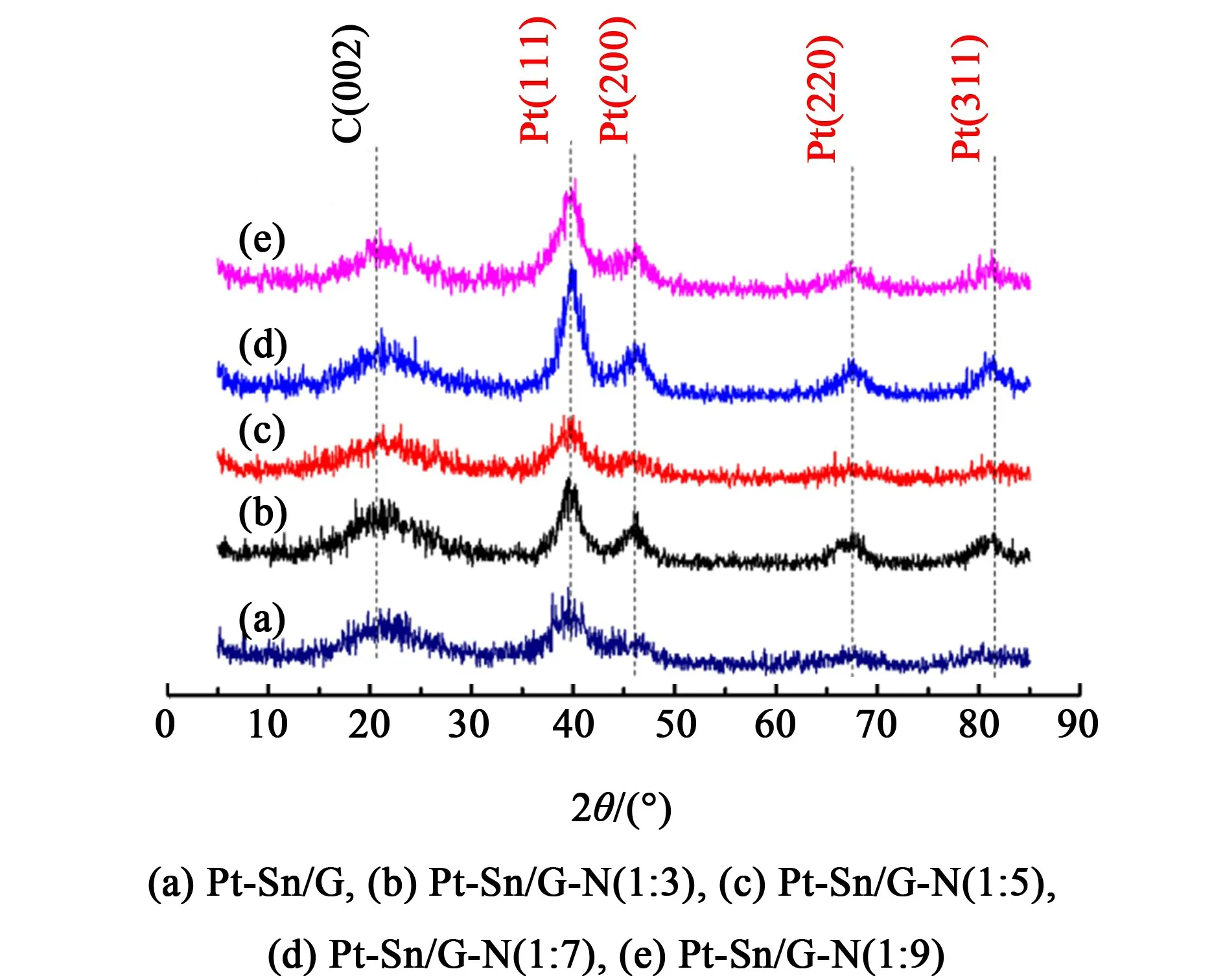
图1 催化剂Pt-Sn/G和Pt-Sn/G-N的XRD衍射图
在2θ为39.7°、46.1°、67.4°和81.4°处的衍射峰分别对应Pt的(111)、(200)、(220)、(311)晶面衍射峰[14],证明Pt颗粒负载到石墨烯上。但是未出现Sn的衍射峰,这可能是因为Sn以氧化物或者与Pt形成合金态的形式存在。通过Scherrer公式[15]:
D=0.94λ/(Bcosθ)
(1)
利用Pt(220)晶面X射线衍射峰估算Pt纳米粒子的粒径,得到样品Pt-Sn/G、Pt-Sn/G-N(1∶3)、Pt-Sn/G-N(1∶5)、Pt-Sn/G-N(1∶7)和Pt-Sn/G-N(1∶9)的粒径依次为3.0、4.5、3.5、2.9和3.8 nm。可得样品 Pt-Sn/G-N(1∶7)的Pt纳米粒子的粒径最小。
图2 样品FT-IR衍射图

催化剂样品的金属Pt和Sn的负载率以及N/C原子数比见表1。
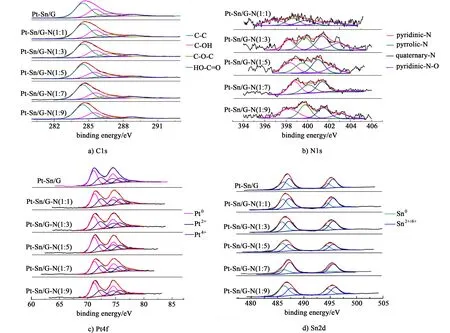
图3 催化剂Pt-Sn/G和Pt-Sn/G-N的X射线光电子能谱a) C1s、b) N1s、c) Pt4f、d) Sn2d的分峰图
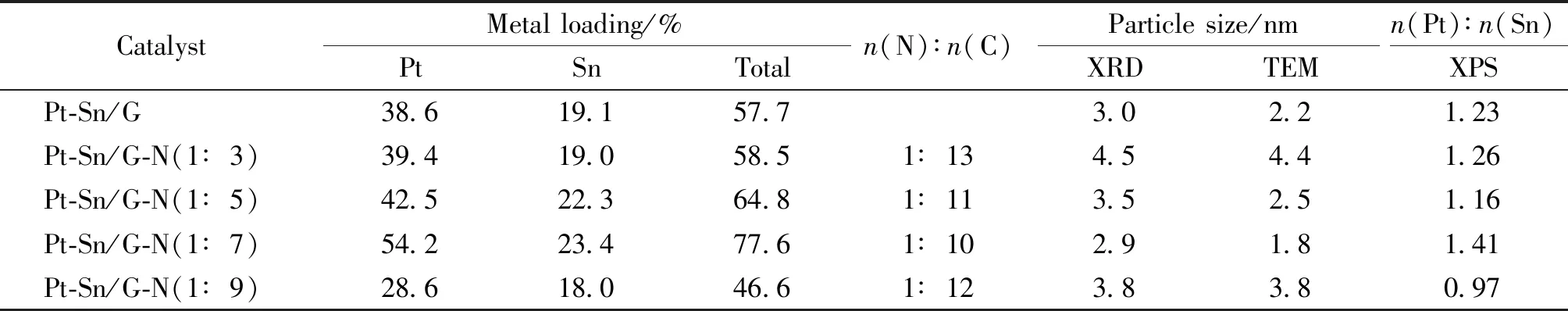
表1 不同催化剂样品的金属负载率、N/C原子数比和金属颗粒尺寸Table 1 Metal loading, N∶C atomic ratios and particle sizes of the catalysts
由表1可以看出催化剂Pt-Sn/G的金属Pt和Sn的负载量与催化剂Pt-Sn/G-N(1∶3)的负载量相差无几,总的金属负载量都在58%左右;在掺氮催化剂中,发现随着水合肼量的增加金属Pt和Sn的负载量开始都增大,到催化剂Pt-Sn/G-N(1∶7)时Pt和Sn的负载量都各自达到最大,总的金属负载量达到77.6%;但随着水合肼的继续增加,金属负载量都急剧减小,总的负载量只有46.6%,这可能是由于水合肼用量过多,一定程度上破坏了石墨烯的结构造成团聚,载体比表面积减小,使得颗粒负载量急剧减小;由表1也发现N与C的原子数比也随着水合肼量的增加先增大后减小,在催化剂Pt-Sn/G-N(1∶7)样品中n(N)∶n(C)最大为1∶10,因此此样品中的载体材料石墨烯的含氮量最高。可以看出含氮量变化与各金属负载量的变化趋势一样,但样品含氮量最大时Pt和Sn的负载量也达到最大,这说明氮元素对金属的负载起到很大的促进作用,这可能是因为N元素的掺杂部位可作为金属颗粒锚定的活性位点,N元素的增加使得活性位点增加,因此有更多的金属颗粒负载到载体材料石墨烯上[21],所以样品Pt-Sn/G-N(1∶7)的金属纳米颗粒的负载率最高。
图4为Pt-Sn/G和不同Pt-Sn/G-N催化剂的TEM图片和其所对应的粒径分布柱状图。图4显示每个样品的纳米颗粒都成功负载到石墨烯上,从图4a)可以看出,未掺氮样品Pt-Sn/G的纳米颗粒分散性较好,但整体负载密度过大,有一定的团聚现象。图4b)可以看出样品Pt-Sn/G-N(1∶3)的纳米颗粒有明显的团聚,粒径分布范围也非常大,粒径尺寸不均匀且比较大。图4c)和图4d)分别为样品Pt-Sn/G-N(1∶5)和Pt-Sn/G-N(1∶7)的TEM图,可以看出2个样品的纳米颗粒分布基本均匀,没有明显的团聚现象,其中样品Pt-Sn/G-N(1∶7)的粒径更小。再由其相应的粒径分布柱状图可以看出样品Pt-Sn/G-N(1∶7)的粒径分布范围更窄,说明样品Pt-Sn/G-N(1∶7)的纳米颗粒尺寸分布更均一。但是,由图4e)可以看出样品Pt-Sn/G-N(1∶9)的纳米颗粒分散性变差,局部有明显的团聚现象且颗粒粒径也比较大。这表明不同量的水合肼还原氧化石墨烯得到的掺氮石墨烯对金属纳米颗粒的分散情况有直接的影响:当GO与水合肼的质量比为1∶7时,催化剂分散更为理想,颗粒粒径最小,平均粒径为1.8 nm,且颗粒近似球形,其催化比表面积显著增大;随着水合肼量的进一步增大,催化剂的颗粒粒径变大,且有明显的团聚现象,因此在不同的Pt-Sn/G-N催化剂中,Pt-Sn/G-N(1∶7)的催化剂颗粒分布最好,并且其相比未掺氮催化剂Pt-Sn/G的纳米颗粒分布均匀疏松,粒径也更小,催化比表面积更大。其各个催化剂的颗粒平均粒径尺寸见表1,可以发现XRD与TEM的测试结果有所偏差,这可能是由于用谢乐公式计算过程中有一定程度的误差,但样品的颗粒尺寸变化趋势一样,都是催化剂Pt-Sn/G-N(1∶7)的粒径最小。
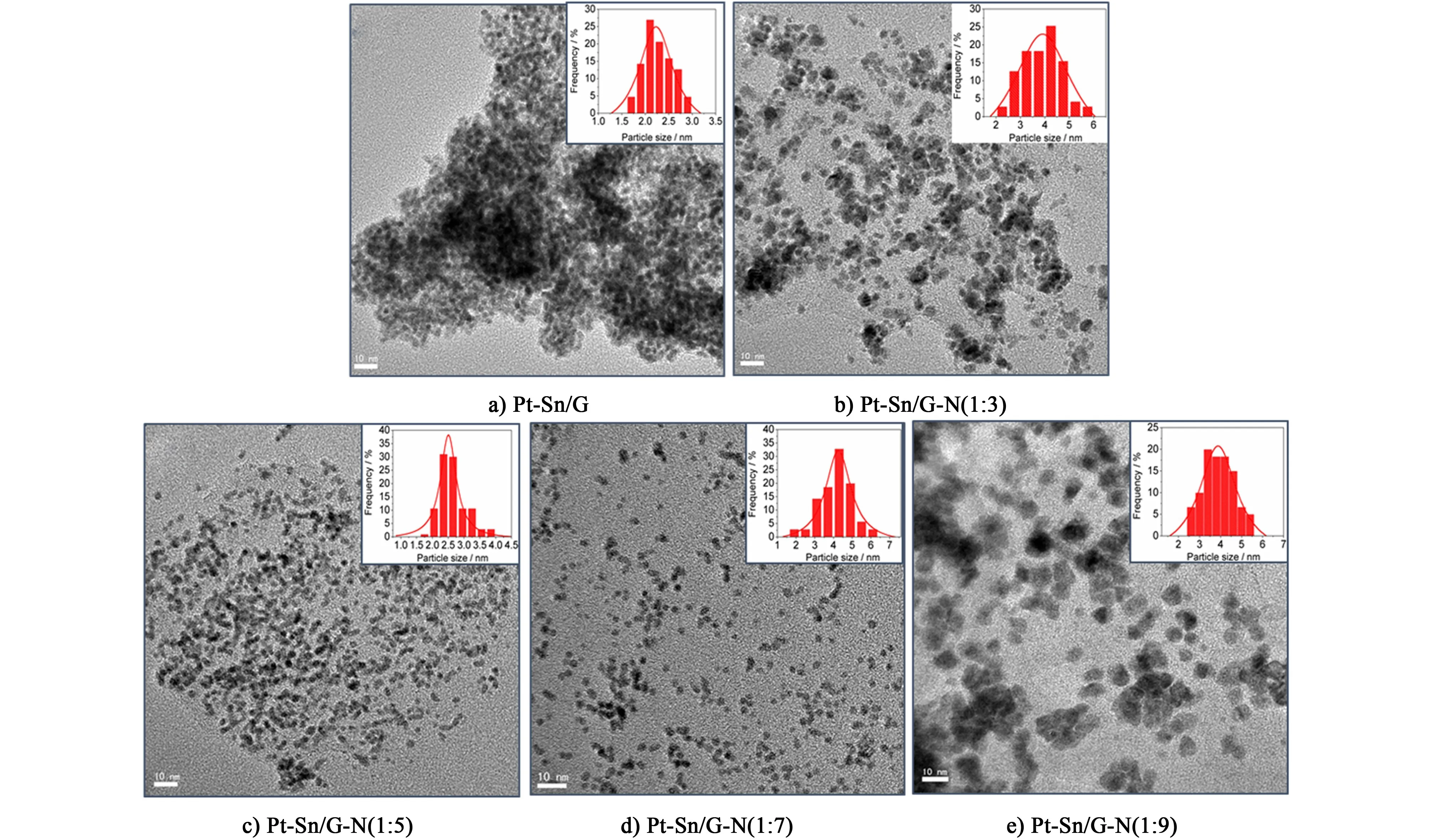
图4 催化剂Pt-Sn/G和Pt-Sn/G-N样品的TEM图片和其对应的粒径分布柱状图
2.2 电化学分析
图5为不同催化剂典型的氢吸附/脱附曲线。
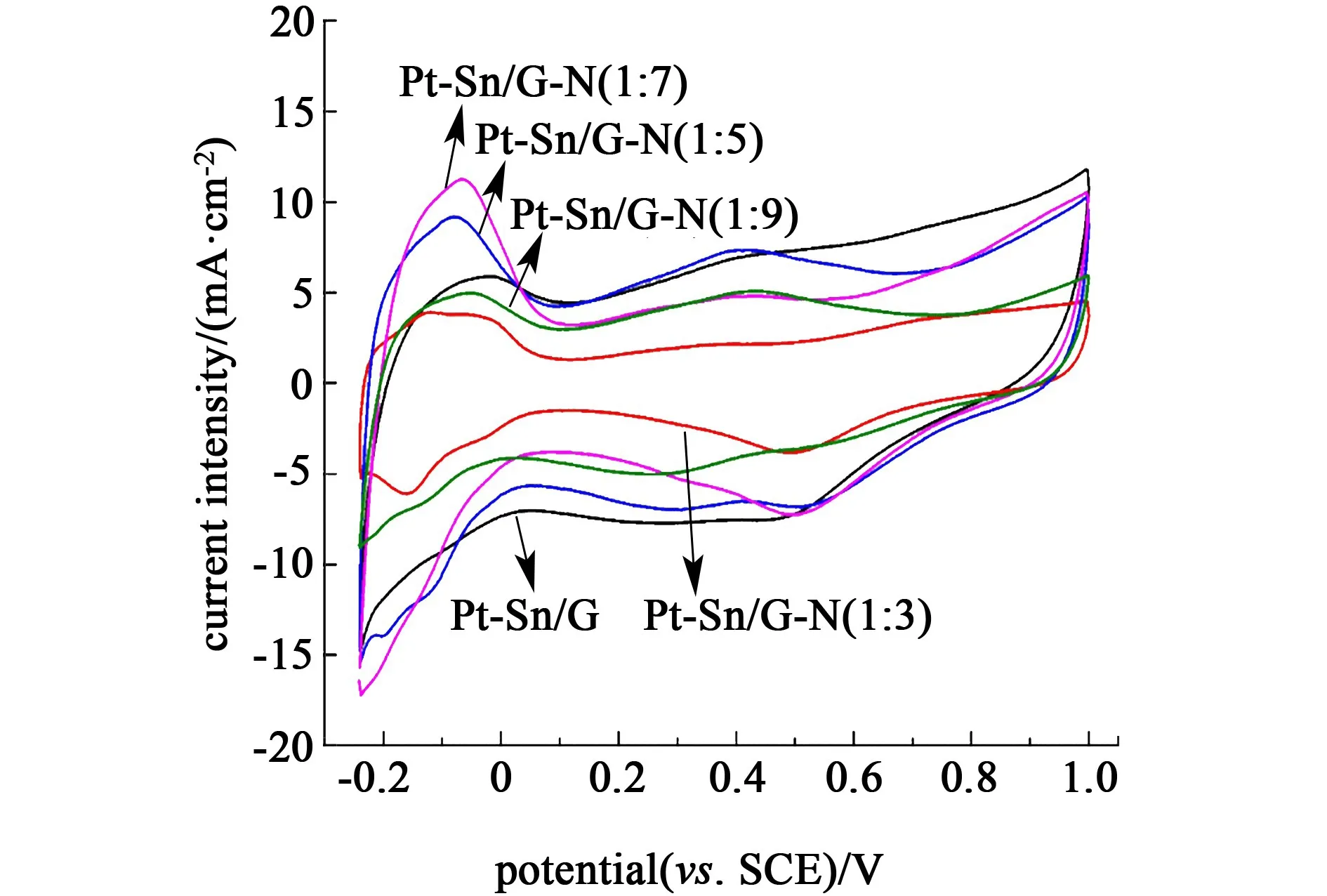
图5 不同样品催化剂在0.5 mol/L 硫酸水溶液中的循环伏安图,扫描速率为50 mV/s
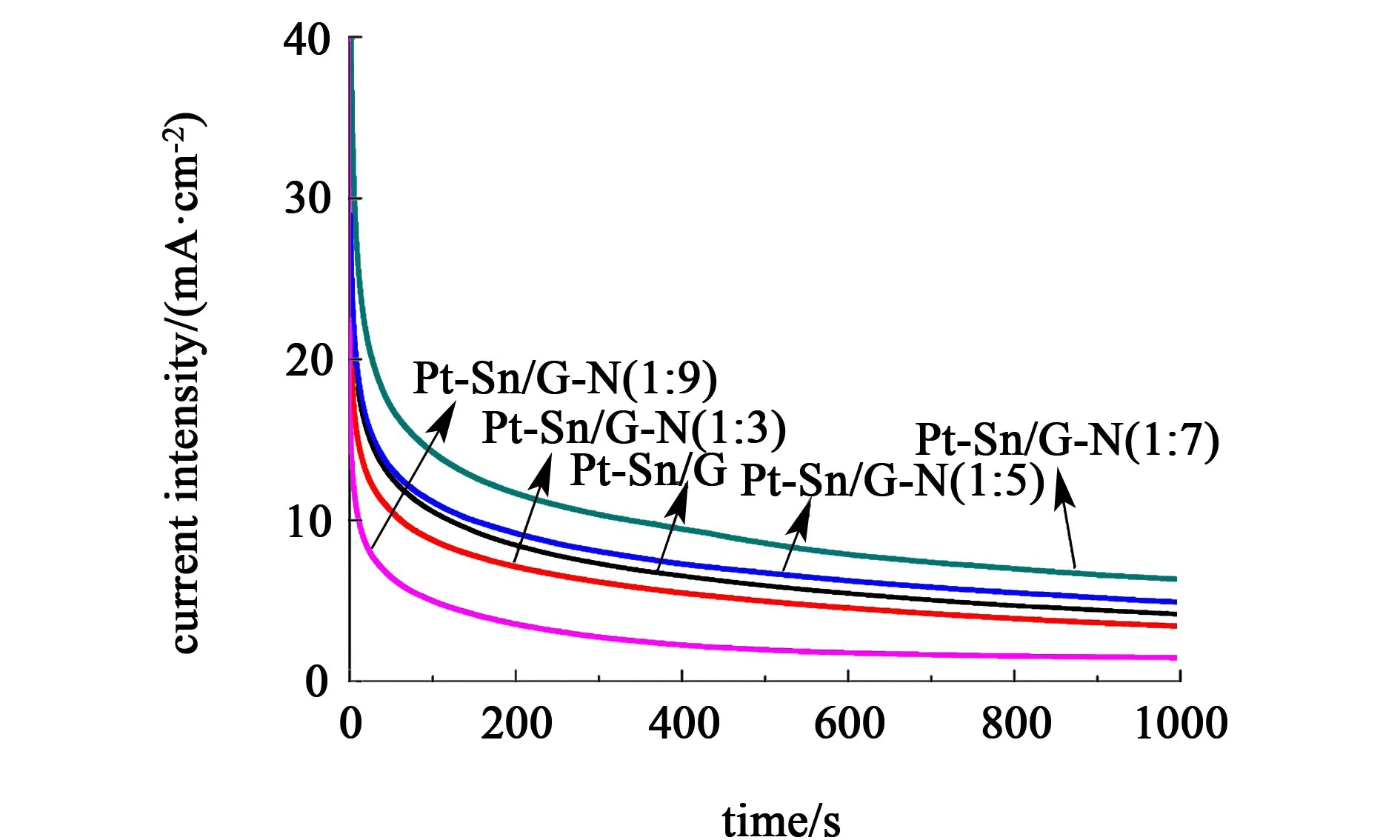
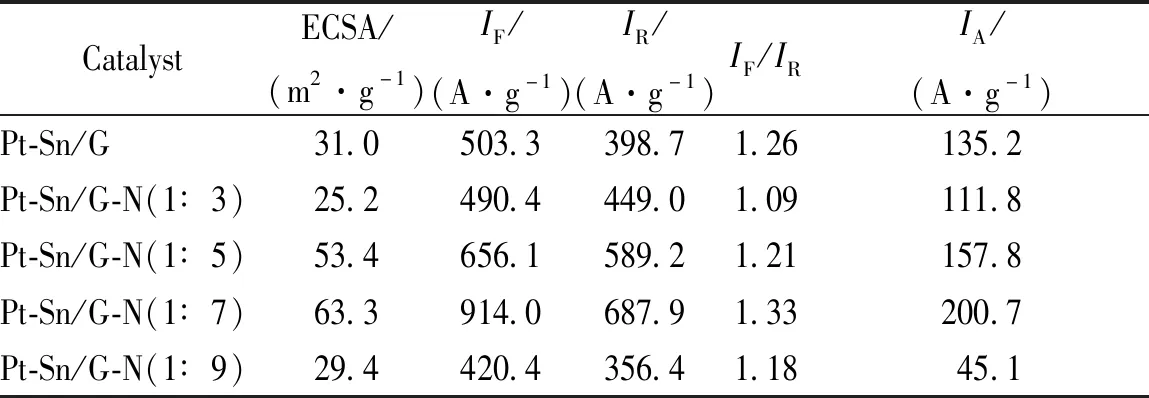
由图5可知样品Pt-Sn/G-N(1∶7)的氢吸附/脱附峰面积明显大于Pt-Sn/G及其他Pt-Sn/G-N样品,具体数值如表2所示。电化学活性面积(ECSA)从小到大依次是Pt-Sn/G-N(1∶3) 图6为催化剂乙醇氧化反应的循环伏安图,正扫过程中出现的特征峰(If)是乙醇的氧化反应峰,反扫过程中的氧化峰(Ir)则是由于在正扫过程中产生的CO等中间产物的解吸附反应峰[23-24]。 图6 不同催化剂在1.0 mol/L的乙醇+0.5 mol/L硫酸溶液中的循环伏安图,扫描速率为20 mV/s 各样品的特征峰值如表2所示,可以发现Pt-Sn/G-N(1∶7)样品的正扫描峰电流密度明显高于其他样品,这正是由于样品的负载量高,提高了催化剂的催化活性。并且正扫峰电流密度与反扫峰的比值可以反应催化剂抗CO中毒性能,比值越大,抗CO中毒性能越高,即对乙醇氧化的活性越高[25]。由表2可知,样品Pt-Sn/G-N(1∶7)的电催化活性最高。 图7为不同催化剂样品在0.5 mol/L H2SO4+1 mol/L CH2CH3OH溶液中的计时电流曲线。 图7 不同催化剂在1.0 mol/L的乙醇+0.5 mol/L硫酸溶液中的计时电流曲线 由图7可以看出,在前50 s所有样品的电流密度都快速地衰减,继而缓慢衰减至稳定的电流密度。电流密度起初快速衰减的原因可能是由于乙醇电催化反应生成的一些中间产物吸附在金属纳米粒子上,还来不及氧化从而导致的催化剂活性位点失活,所引起的电流密度快速下降。1 000 s以后的电流密度值见表2,大小依次为Pt-Sn/G-N(1∶7)>Pt-Sn/G-N(1∶5)>Pt-Sn/G>Pt-Sn/G-N(1∶3)>Pt-Sn/G-N(1∶9)。说明样品Pt-Sn/G-N(1∶7)对乙醇的电氧化具有更稳定的极化电流,这结果与电化学活性面积大小的结果一致。 表2 不同催化剂样品的电催化活性参数Table 2 Electrocatalytic activity parameters of the Pt-Sn/G and Pt-Sn/G-N catalysts 使用一系列不同量的水合肼作为氮源,并还原氧化石墨烯,再负载Pt-Sn金属粒子,制备Pt-Sn/G-N复合材料作为电极催化剂。与乙二醇还原制得的Pt-Sn/G催化剂作对比,考察了不同量的水合肼还原氧化石墨烯对催化剂结构、组分及性能的影响。研究表明水合肼含量对金属颗粒含量有显著影响,石墨烯中氮含量随水合肼含量的增加而增加,当石墨烯与水合肼的质量比为1∶7时,氮元素含量最高,负载了更多均一且尺寸较小的晶粒,同时具有最高的电催化活性、抗CO中毒性能和稳定性。但随着水合肼含量的进一步增加,Pt-Sn金属颗粒团聚,催化活性性能下降。因此,通过适当控制含氮量的掺入,得到了一种具有优良催化活性的电催化剂,Pt与Sn的协同作用效果最佳,并在直接乙醇燃料电池(DEFCs)中具有实际应用前景。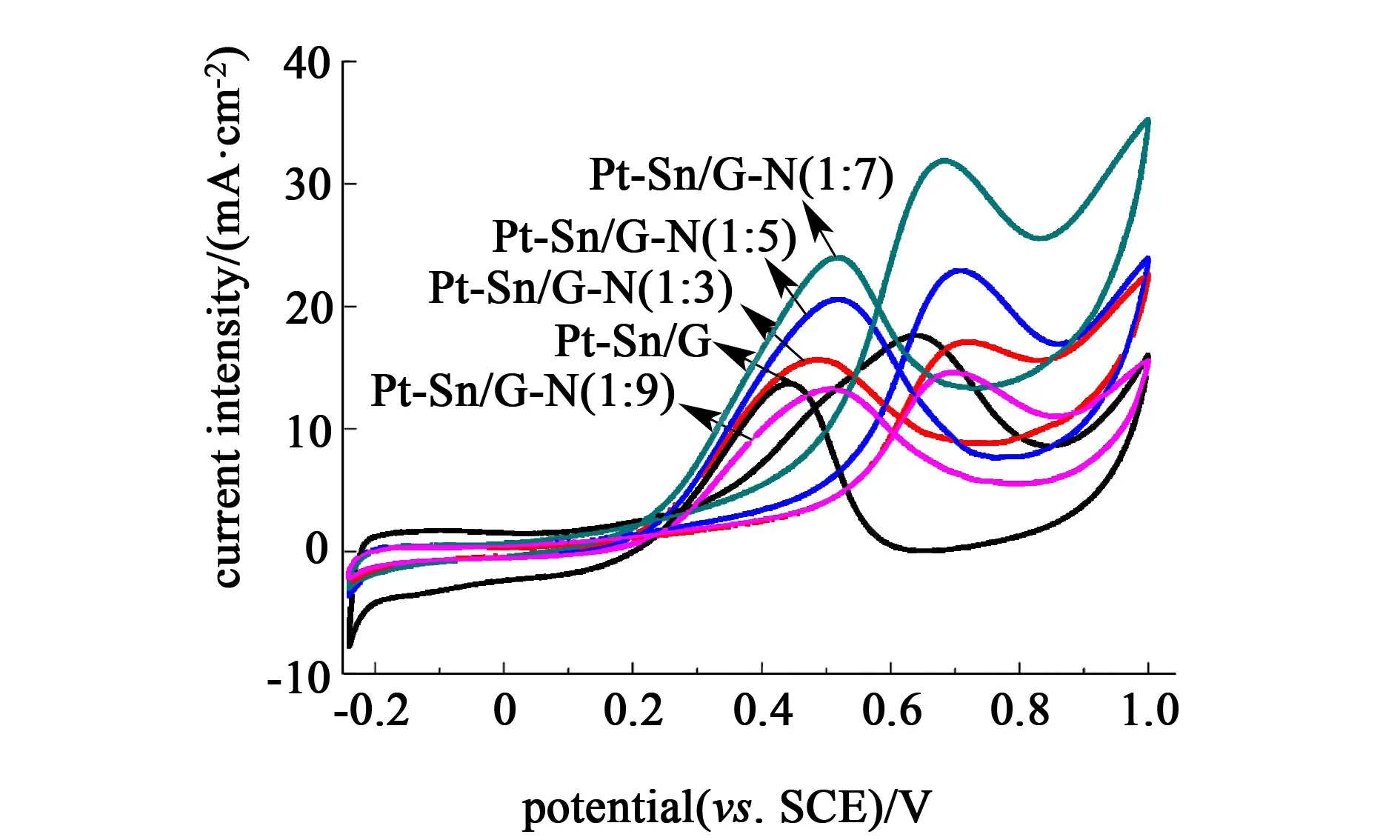
3 结论
猜你喜欢
化学工程师(2023年1期)2023-02-17 15:09:48
氯碱工业(2022年6期)2022-11-21 01:41:32
化工环保(2021年2期)2021-04-25 13:42:34
理化检验-化学分册(2020年12期)2021-01-26 00:41:38
中国果树(2020年2期)2020-07-25 02:14:28
云南化工(2020年5期)2020-06-12 09:26:40
盐科学与化工(2019年11期)2019-12-04 02:16:42
上海农业科技(2019年1期)2019-02-22 01:51:28
淮南师范学院学报(2015年3期)2015-03-22 01:16:19
河北科技大学学报(2015年5期)2015-03-11 16:16:34
